
Abstract
Background
Long noncoding RNA (lncRNA) is generally identified as competing endogenous RNA (ceRNA) that plays a vital role in the pathogenesis of kidney renal clear cell carcinoma (KIRC), the most common subtype of renal cell carcinoma with poor prognosis and unclear pathogenesis. This study established a novel ceRNA network and thus identified a three-lncRNA prognostic model in KIRC patients.
Methods
Differentially expressed genes (DEGs) were screened out from The Cancer Genome Atlas (TCGA) database. The lncATLAS was applied to determine the differentially expressed lncRNAs (DElncRNAs) of the cytoplasm. The miRcode, miRDB, miRTarBase, and TargetScan databases were utilized to predict the interactions of DElncRNAs, DEmiRNAs, and DEmRNAs. Cytoscape was used to construct the ceRNA network. Then, a lncRNA prognostic model (LPM) was constructed based on ceRNA-related lncRNA that was significantly related to overall survival (OS), and its predictive ability was evaluated. Moreover, an LPM-based nomogram model was constructed. The significantly different expression of genes in the LPM was validated in an independent clinical cohort (N = 21) by quantitative RT-PCR.
Results
A novel ceRNA regulatory network, including 73 lncRNAs, 8 miRNAs, and 21 mRNAs was constructed. Functional enrichment analysis indicated that integral components of membrane and PI3K–Akt signaling pathway represented the most significant GO terms and pathway, respectively. The LPM established based on three lncRNAs (MIAT, LINC00460, and LINC00443) of great prognostic value from the ceRNA network was proven to be independent of conventional clinical parameters to differentiate patients with low or high risk of poor survival, with the AUC of 1-, 5- and 10-year OS were 0.723, 0.714 and 0.826 respectively. Furthermore, the nomogram showed a better predictive value in KIRC patients than individual prognostic parameters. The expression of MIAT and LINC00460 was significantly upregulated in the KIRC samples, while the expression of LINC00443 was significantly downregulated compared with the adjacent normal samples in the clinical cohort, TCGA, and GTEx.
Conclusion
This LPM based on three-lncRNA could serve as an independent prognostic factor with a tremendous predictive ability for KIRC patients, and the identified novel ceRNA network may provide insight into the prognostic biomarkers and therapeutic targets of KIRC.
Similar content being viewed by others


Background
Kidney renal clear cell carcinoma (KIRC) is the most common and aggressive malignant subtype of renal cell carcinoma that has a poor prognosis and high mortality in an advanced stage due to the lack of useful biomarkers and treatments [1]. Currently, there is a multitude of established treatments for KIRC, such as surgical resection, nonspecific immune approach, targeted therapy against vascular endothelial growth factor, and novel immunotherapy agents. Despite these treatments, about 50% of KIRC patients develop metastases, and the 5-year survival rate of these patients is still lower than 10% [2]. At present, the commonly used clinical prognostic markers of KIRC include the pathological grade system and tumor node metastasis (TNM) stage, microvascular invasion, tumor necrosis, and invasion of the collecting system [3]. These clinicopathological risk factors exhibit valuable but insufficient prediction of prognosis and estimation for subsets of KIRC patients. Previous researches have established some prognostic models and nomograms that incorporate necrosis, blood tests such as lactate dehydrogenase, hemoglobin, platelets, and calcium levels, prior nephrectomy, symptoms, and performance status [https://www.gencodegenes.org/). Then, the gene symbols were annotated based on the Homo_sapiens.GRCh38.84.chr.gtf file, which was downloaded from the Ensembl database (https://asia.ensembl.org/index.html).
Identification of differentially expressed genes (DEGs)
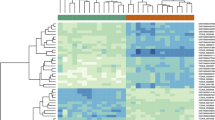
We compared the KIRC samples and adjacent normal samples to identify DEGs by using DESeq 2 R package (Version 1.27.19; http://www.bioconductor.org/packages/devel/bioc/html/DESeq2.html) with a rigorous threshold as |log2-fold change (FC)| > 2.0 and FDR < 0.01 [21]. Then a heat map and volcano plot were drawn by using the heatmap R package (Version 1.0.1; https://www.rdocumentation.org/packages/pheatmap) and ggpubr R package (Version 0.2.4; https://www.rdocumentation.org/packages/ggpubr) in R software (Version 3.6.0; https://www.r-project.org/), to visualize the hierarchical clustering analysis of the identified DEGs.
Construction of the ceRNA network
The lncATLAS database (http://lncatlas.crg.eu/) was used to identify the DElncRNAs located in the cytoplasm [22]. Then the DEmiRNAs which potentially interacted with DElncRNAs located in the cytoplasm were predicted using the miRcode (http://www.mircode.org/), a comprehensive searchable map of putative microRNA target sites in the long noncoding transcriptome [23]. Subsequently, the target DEmRNAs of DEmiRNA were predicted using miRDB (http://mirdb.org/), miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) and TargetScan (http://www.targetscan.org/vert_72/) databases [24,25,26]. After that, Cytoscape software (Version 3.7.2; http://www.cytoscape.org/) was utilized to visualize and construct the ceRNA network [27].
Functional enrichment analysis
The pathway and functional enrichment analysis were carried out by utilizing KO-Based Annotation System (KOBAS) (Version 3.0; http://kobas.cbi.pku.edu.cn/) and the Database for Annotation, Visualization and Integrated Discovery (DAVID) (Version: 6.8; https://david.ncifcrf.gov/), to investigate the potential biological implications of the ceRNA network [ In conclusion, we successfully constructed a novel ceRNA regulatory network, which narrowed the scope of predicting prognostic biomarkers and therapeutic targets for KIRC. Besides, we identified and validated an LPM which is based on three lncRNAs involved in the ceRNA network, and it has independent and great prognostic value for KIRC patients.Conclusions
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional information files.
Abbreviations
- KIRC:
-
Kidney renal clear cell carcinoma
- lncRNA:
-
Long noncoding RNA
- ceRNA:
-
Competitive endogenous RNA
- DElncRNAs:
-
Differentially expressed lncRNAs
- LPM:
-
lncRNA prognostic model
- OS:
-
Overall survival
- TCGA:
-
The Cancer Genome Atlas
- LASSO:
-
Least absolute shrinkage and selection operator
- K-M:
-
Kaplan–Meier
- ROS:
-
Receiver operating characteristic
- DSS:
-
Disease-specific survival
- C-index:
-
Concordance index
- DCA:
-
Decision curve analysis
- GSEA:
-
Gene set enrichment analysis
- GTEx:
-
Genotype-Tissue Expression
- GEPIA2:
-
Gene Expression Profiling Interactive Analysis
- CI:
-
Confidence interval
- AUC:
-
Area under the ROC curve
- HR:
-
Hazard ratio
References
Capitanio U, Montorsi F. Renal cancer. Lancet. 2016;387(10021):894–906. https://doi.org/10.1016/s0140-6736(15)00046-x.
Barata PC. Treatment of renal cell carcinoma: current status and future directions. CA Cancer J Clin. 2017;67:507–24.
Ljungberg B, Albiges L, Abu-Ghanem Y, Bensalah K, Dabestani S, Fernández-Pello S, et al. European association of urology guidelines on renal cell carcinoma: the 2019 update. Eur Urol. 2019;75(5):799–810. https://doi.org/10.1016/j.eururo.2019.02.011.
Heng DY, **e W, Regan MM, Warren MA, Golshayan AR, Sahi C, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol. 2009;27(34):5794–9. https://doi.org/10.1200/jco.2008.21.4809.
Frank I, Blute ML, Cheville JC, Lohse CM, Weaver AL, Zincke H. An outcome prediction model for patients with clear cell renal cell carcinoma treated with radical nephrectomy based on tumor stage, size, grade and necrosis: the SSIGN score. J Urol. 2002;168(6):2395–400. https://doi.org/10.1097/01.ju.0000035885.91935.d5.
Gulati S, Martinez P, Joshi T, Birkbak NJ, Santos CR, Rowan AJ, et al. Systematic evaluation of the prognostic impact and intratumour heterogeneity of clear cell renal cell carcinoma biomarkers. Eur Urol. 2014;66(5):936–48. https://doi.org/10.1016/j.eururo.2014.06.053.
Prensner JR, Chinnaiyan AM. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011;1(5):391.
Lin C, Yang L. Long noncoding RNA in cancer: wiring signaling circuitry. Trends Cell Biol. 2018;28(4):287–301. https://doi.org/10.1016/j.tcb.2017.11.008.
Schmitt AM, Chang HY. Long noncoding RNAs in cancer pathways. Cancer Cell. 2016;29(4):452–63. https://doi.org/10.1016/j.ccell.2016.03.010.
Deng M, Blondeau JJ, Schmidt D, Perner S, Muller SC, Ellinger J. Identification of novel differentially expressed lncRNA and mRNA transcripts in clear cell renal cell carcinoma by expression profiling. Genom Data. 2015;5:173–5. https://doi.org/10.1016/j.gdata.2015.06.016.
Blondeau JJ, Deng M, Syring I, Schrodter S, Schmidt D, Perner S, et al. Identification of novel long non-coding RNAs in clear cell renal cell carcinoma. Clin Epigenetics. 2015;7:10. https://doi.org/10.1186/s13148-015-0047-7.
Fachel AA, Tahira AC, Vilella-Arias SA, Maracaja-Coutinho V, Gimba ER, Vignal GM, et al. Expression analysis and in silico characterization of intronic long noncoding RNAs in renal cell carcinoma: emerging functional associations. Mol Cancer. 2013;12(1):140. https://doi.org/10.1186/1476-4598-12-140.
Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17(1):47–62. https://doi.org/10.1038/nrg.2015.10.
Chen LL. Linking long noncoding RNA localization and function. Trends Biochem Sci. 2016;41(9):761–72. https://doi.org/10.1016/j.tibs.2016.07.003.
Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell. 2013;152(6):1298–307. https://doi.org/10.1016/j.cell.2013.02.012.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146(3):353–8. https://doi.org/10.1016/j.cell.2011.07.014.
Cesana M, Cacchiarelli D, Legnini I, Santini T, Sthandier O, Chinappi M, et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147(2):358–69. https://doi.org/10.1016/j.cell.2011.09.028.
Wang L, Cho KB, Li Y, Tao G, **e Z, Guo B. Long noncoding RNA (lncRNA)-mediated competing endogenous rna networks provide novel potential biomarkers and therapeutic targets for colorectal cancer. Int J Mol Sci. 2019. https://doi.org/10.3390/ijms20225758.
Afify AY, Ibrahim SA, Aldamsisi MH, Zaghloul MS, El-Ekiaby N, Abdelaziz AI. Competing endogenous RNAs in hepatocellular carcinoma-the pinnacle of rivalry. Semin Liver Dis. 2019;39(4):463–75. https://doi.org/10.1055/s-0039-1688442.
Yang J, Qiu Q, Qian X, Yi J, Jiao Y, Yu M, et al. Long noncoding RNA LCAT1 functions as a ceRNA to regulate RAC1 function by sponging miR-4715-5p in lung cancer. Mol Cancer. 2019;18(1):171. https://doi.org/10.1186/s12943-019-1107-y.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Mas-Ponte D, Carlevaro-Fita J, Palumbo E, Hermoso Pulido T, Guigo R, Johnson R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA. 2017;23(7):1080–7. https://doi.org/10.1261/rna.060814.117.
Jeggari A, Marks DS, Larsson E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics. 2012;28(15):2062–3. https://doi.org/10.1093/bioinformatics/bts344.
Wong N, Wang X. miRDB: an online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2015;43(Database issue):D146–52. https://doi.org/10.1093/nar/gku1104.
Chou CH, Chang NW, Shrestha S, Hsu SD, Lin YL, Lee WH, et al. miRTarBase 2016: updates to the experimentally validated miRNA-target interactions database. Nucleic Acids Res. 2016;44(D1):D239–47. https://doi.org/10.1093/nar/gkv1258.
Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015. https://doi.org/10.7554/elife.05005.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. https://doi.org/10.1101/gr.1239303.
**e C, Mao X, Huang J, Ding Y, Wu J, Dong S, et al. KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011;39(Web Server issue):W316–22. https://doi.org/10.1093/nar/gkr483.
Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4(5):P3.
Long J, Wang A, Bai Y, Lin J, Yang X, Wang D, et al. Development and validation of a TP53-associated immune prognostic model for hepatocellular carcinoma. EBioMedicine. 2019;42:363–74. https://doi.org/10.1016/j.ebiom.2019.03.022.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res. 2004;10(21):7252–9. https://doi.org/10.1158/1078-0432.Ccr-04-0713.
Kiran M, Chatrath A, Tang X, Keenan DM, Dutta A. A prognostic signature for lower grade gliomas based on expression of long non-coding RNAs. Mol Neurobiol. 2019;56(7):4786–98. https://doi.org/10.1007/s12035-018-1416-y.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. https://doi.org/10.1073/pnas.0506580102.
Tang Z, Kang B, Li C, Chen T, Zhang Z. GEPIA2: an enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019;47(W1):W556–60. https://doi.org/10.1093/nar/gkz430.
Jiang W, Guo Q, Wang C, Zhu Y. A nomogram based on 9-lncRNAs signature for improving prognostic prediction of clear cell renal cell carcinoma. Cancer Cell Int. 2019;19:208. https://doi.org/10.1186/s12935-019-0928-5.
Deng X, Lin D, Chen B, Zhang X, Xu X, Yang Z, et al. Development and validation of an IDH1-associated immune prognostic signature for diffuse lower-grade glioma. Front Oncol. 2019;9:1310. https://doi.org/10.3389/fonc.2019.01310.
Vickers AJ, Elkin EB. Decision curve analysis: a novel method for evaluating prediction models. Med Decis Making. 2006;26(6):565–74. https://doi.org/10.1177/0272989x06295361.
Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, et al. Transcriptomic profiling of the tumor microenvironment reveals distinct subgroups of clear cell renal cell cancer: data from a randomized phase III trial. Cancer Discov. 2019;9(4):510–25. https://doi.org/10.1158/2159-8290.Cd-18-0957.
Barbieri CE, Chinnaiyan AM, Lerner SP, Swanton C, Rubin MA. The emergence of precision urologic oncology: a collaborative review on biomarker-driven therapeutics. Eur Urol. 2017;71(2):237–46. https://doi.org/10.1016/j.eururo.2016.08.024.
Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–46. https://doi.org/10.1038/nature10887.
Zhou S, Treloar AE, Lupien M. Emergence of the noncoding cancer genome: a target of genetic and epigenetic alterations. Cancer Discov. 2016;6(11):1215–29. https://doi.org/10.1158/2159-8290.Cd-16-0745.
Reis-Filho JS, Pusztai L. Gene expression profiling in breast cancer: classification, prognostication, and prediction. Lancet. 2011;378(9805):1812–23. https://doi.org/10.1016/s0140-6736(11)61539-0.
Zhang C, Huang D, Liu A, Xu Y, Na R, Xu D. Genome-wide screening and cohorts validation identifying novel lncRNAs as prognostic biomarkers for clear cell renal cell carcinoma. J Cell Biochem. 2020;121(3):2559–70. https://doi.org/10.1002/jcb.29478.
Cao Z, Pan X, Yang Y, Huang Y, Shen HB. The lncLocator: a subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics. 2018;34(13):2185–94. https://doi.org/10.1093/bioinformatics/bty085.
Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet. 2013;45(8):860–7. https://doi.org/10.1038/ng.2699.
Guo H, German P, Bai S, Barnes S, Guo W, Qi X, et al. The PI3K/AKT pathway and renal cell carcinoma. J Genet Genomics. 2015;42(7):343–53. https://doi.org/10.1016/j.jgg.2015.03.003.
Zeng JH, Lu W, Liang L, Chen G, Lan HH, Liang XY, et al. Prognosis of clear cell renal cell carcinoma (ccRCC) based on a six-lncRNA-based risk score: an investigation based on RNA-sequencing data. J Transl Med. 2019;17(1):281. https://doi.org/10.1186/s12967-019-2032-y.
Liu H, Ye T, Yang X, Lv P, Wu X, Zhou H, et al. A panel of four-lncRNA signature as a potential biomarker for predicting survival in clear cell renal cell carcinoma. J Cancer. 2020;11(14):4274–83. https://doi.org/10.7150/jca.40421.
Li K, Sun D, Gou Q, Ke X, Gong Y, Zuo Y, et al. Long non-coding RNA linc00460 promotes epithelial-mesenchymal transition and cell migration in lung cancer cells. Cancer Lett. 2018;420:80–90. https://doi.org/10.1016/j.canlet.2018.01.060.
Liu X, Wen J, Wang H, Wang Y. Long non-coding RNA LINC00460 promotes epithelial ovarian cancer progression by regulating microRNA-338-3p. Biomed Pharmacother. 2018;108:1022–8. https://doi.org/10.1016/j.biopha.2018.09.103.
Feng L, Yang B, Tang XD. Long noncoding RNA LINC00460 promotes carcinogenesis via sponging miR-613 in papillary thyroid carcinoma. J Cell Physiol. 2019;234(7):11431–9. https://doi.org/10.1002/jcp.27799.
Qu Y, **ao H, **ao W, **ong Z, Hu W, Gao Y, et al. Upregulation of MIAT regulates LOXL2 expression by competitively binding MiR-29c in clear cell renal cell carcinoma. Cell Physiol Biochem. 2018;48(3):1075–87. https://doi.org/10.1159/000491974.
Wang J, Zhang C, He W, Gou X. Construction and comprehensive analysis of dysregulated long non-coding RNA-associated competing endogenous RNA network in clear cell renal cell carcinoma. J Cell Biochem. 2018. https://doi.org/10.1002/jcb.27557.
Yang K, Lu XF, Luo PC, Zhang J. Identification of six potentially long noncoding RNAs as biomarkers involved competitive endogenous RNA in clear cell renal cell carcinoma. Biomed Res Int. 2018;2018:9303486. https://doi.org/10.1155/2018/9303486.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
ZD initiated the study and organized; ZD, ZS and Hu XP designed and carried out bioinformatics analyses, statistical analyses, drew figures and drafted the manuscript; ZS and Hu XP participated in modifying the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was carried out in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). All patients signed the informed consent, and this study was approved by the ethics committees of Bei**g Chao-Yang Hospital.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
List of primers used for RT-PCR. Table S2. Differentially expressed lncRNAs, miRNAs, and mRNAs between KIRC samples and adjacent normal samples. Table S3. The subcellular distribution of lncRNAs in the ceRNA network. Table S4. The interactions of the ceRNA network in KIRC. Table S5. Seventeen lncRNAs associated with overall survival in KIRC. Table S6. The results of GSEA. Table S7. The potential binding sites of three lncRNAs (LINC00443, LINC00460 and MIAT) on the targeted DEmiRNAs in the ceRNA network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, D., Zeng, S. & Hu, X. Identification of a three-long noncoding RNA prognostic model involved competitive endogenous RNA in kidney renal clear cell carcinoma. Cancer Cell Int 20, 319 (2020). https://doi.org/10.1186/s12935-020-01423-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01423-4