
Abstract
Background
Metabolic syndrome (MetS) is characterized by central obesity, insulin resistance, dysglycemia, and a pro-atherogenic plasma lipid profile. MetS creates a high risk for development of type 2 diabetes (T2DM) and cardiovascular disease (CVD), presumably by altering inflammatory responses. Presently, it is unknown how the chronic metabolic disturbances in acute hyperglycemia, MetS and T2DM affect the immune activity of peripheral blood cells.
Methods
We performed genome-wide expression analysis of peripheral blood cells obtained from patients with T2DM (n = 6) and age-, sex- , BMI- and blood pressure-matched obese individuals with MetS (n = 4) and lean healthy normoglycemic controls (n = 3), both under fasting conditions and after controlled induction of acute hyperglycemia during a 70 min hyperglycemic clamp. Differential gene expression during fasting conditions was confirmed by real-time PCR, for which we included additional age-, sex-, BMI-, and blood pressure-matched obese individuals with (n = 4) or without (n = 4) MetS.
Results
Pathway and Gene ontology analysis applied to baseline expression profiles of peripheral blood cells from MetS and T2DM patients revealed metabolic changes, highly similar to a reoviral infection gene signature in T2DM patients. Transcription factor binding site analysis indicated that increased HIF-1α activity, a transcription factor induced by either hypoxia or oxidative stress, is responsible for this aberrant metabolic profile in peripheral blood cells from T2DM patients. Acute hyperglycemia in healthy controls resulted in reduced expression of cytotoxicity-related genes, representing NK- and CD8+ cells. In obese controls, MetS and especially T2DM patients, baseline expression of genes involved in cytotoxicity was already low, compared to healthy controls and did not further decrease upon acute hyperglycemia.
Conclusions
The reduced activity of cytotoxic genes in T2DM is explained by chronic hyperglycemia, but its acute effects are restricted to healthy controls. Genome expression of circulating leukocytes from T2DM patients differs from MetS individuals by a specific reovirus signature. Our data thus suggest a role for suppressed anti-viral capacity in the etiology of diabetes.
Similar content being viewed by others


Background
The incidence of metabolic syndrome (MetS) and type 2 diabetes (T2DM) with its associated mortality and morbidity is rapidly increasing in the western world, leading to an extensive medical and societal burden. MetS is defined by a complex set of clinical parameters, which all constitute risk factors for the development of T2DM. These risk factors include central obesity, dyslipidemia (raised triglycerides and lowered high-density lipoprotein cholesterol), high blood pressure, and elevated fasting plasma glucose levels [1].
High blood pressure, hyperglycemia and dislypidemia are held responsible for the increased risk for cardiovascular disease in MetS and T2DM patients, including microvascular complications (including retinopathy, nephropathy and neuropathy) and atherosclerosis at the macrovascular level [1–4]. Both high fasting glucose levels and impaired glucose tolerance are associated with increased cardiovascular events [5], while impaired glucose tolerance predicts cardiovascular death [6]. Intensive glucose control, however, only modestly reduces cardiovascular events [3, 6], indicating a more systemic dysregulation in these patients.
T2DM may develop after a stage of insulin resistance in subjects with MetS. However, only approximately one-third of obese, insulin-resistant individuals develop T2DM because of an inability of beta cells to produce sufficient amounts of insulin [7]. Systemic and local activation of the immune system accompanies obesity, and contributes to the development of insulin resistance, T2DM and cardiovascular disease [8, 9]. It is not entirely understood which mechanisms trigger the onset of T2DM in subjects at risk, but inflammation is a critical candidate. Pancreatic inflammation in T2DM has been shown by increased local infiltration of macrophages in the beta cell areas [10, 11]. In animal models for type 2 diabetes, characterization of the increased infiltrating islet-associated macrophages indicated a pro-inflammatory M1 phenotype, with production of IL-1β and TNFα [12]. Adipose tissue may become a source of inflammation as well, as adipocyte hypertrophy is associated with increased macrophage accumulation, which produce proinflammatory mediators such as TNFα and IL-6, in obese individuals [8]. Trials aimed at inhibition of the immune system by blockade of IL-1α and –β signaling (anakinra) and inhibition of NFκB have shown to reduce HbA1c levels suggesting an effect on beta cell function in patients with T2DM [7, 12]. In animal models, the IL-1 receptor antagonist reduced the numbers of macrophages in islet areas and improved insulin sensitivity and beta cell function [12]. Infiltrating circulating immune cells are thus important in the development of islet dysfunction, in inflammatory adipose tissue, and in the development of atherosclerotic plaques leading to macrovascular disease. The consequence of acute hyperglycemia on circulating immune cells is largely unknown, but a proinflammatory role for hyperglycemia has been observed, because an oral glucose tolerance test increases transcript levels for ICAM-1, TNFα, and IL-6 in peripheral white blood cells from MetS subjects, but not from healthy controls without MetS [13].
Thus blocking inflammation improves glucose tolerance in T2DM patients, but whether in vivo hyperglycemia is able to initiate the activation of the immune system, without the influence of obesity, dyslipidemia, and high blood pressure, is not known. Along this line, it is thus also unknown whether the immune response to acute hyperglycemia is different between healthy subjects and MetS subject and T2DM patients.
Therefore we performed genome-wide expression profiling to achieve an unbiased analysis of the effect of hyperglycemia and T2DM on circulating immune cells. Peripheral blood cells express a large proportion (approximately 80 %) of the genes encoded in the human genome [14]. In addition, the changes in the expression levels of individual genes reflects changes in the (micro)environment of peripheral blood cells and may also reflect organ-specific changes to the same milieu [14], thus serving as a diagnostic tool for a diseased state. Previous studies using peripheral blood profiling revealed that in T2DM patients the mitochondrial oxidative phosphorylation pathway was affected compared to younger lean controls [15]. This finding is remarkable as a reduced expression of oxidative phosphorylation genes was previously established in muscle tissue from T2DM patients [16]. The immune system is affected by metabolic changes in various organs, and vice versa, the immune system influences metabolic parameters such as insulin resistance. Because of this cross-talk the immune system plays a central role in metabolic changes. To be able to obtain a global and unbiased overview of the changes in the immune system, in response to hyperglycemia and T2DM, we studied the alterations in gene expression in whole blood in T2DM patients in comparison with age-, sex- blood pressure- and BMI- matched MetS subjects and lean controls during fasting conditions and after a well-controlled hyperglycemic clamp. We reasoned that this approach may lead to an improved understanding of the underlying pathology of T2DM and may give leads for medical interventions. The comparison of our data with expression data sets from other studies under various experimental conditions, using pathway analysis, allowed us to identify novel pathological pathways.
Methods
Patients and controls
Individuals with MetS and patients with T2DM were recruited by advertisements in local newspapers and studied at the Clinical Research Unit of the Diabetes Center, Department of Internal Medicine at the VU University Medical Center (VUMC). MetS was defined according to the criteria of the International Diabetes Federation (IDF) [17]. In short, subjects were eligible when they had a waist of ≥ 94 cm or more and additionally, two or more of the following criteria: fasting triglycerides ≥ 1.7 mmol/L, HDL cholesterol < 1.03 mmol/L, blood pressure >130/85 mmHg (average of three measurements) or treatment of previously diagnosed hypertension; fasting plasma glucose level (FPG) ≥ 5.6 and < 7.1 mmol/L.
The response to acute elevations of blood glucose levels in T2DM (n = 6), MetS (n = 4) and controls (n = 3) was measured during the hyperglycemic portion of a combined euglycemic-hyperinsulinemic and hyperglycemic-arginine clamp, as described earlier [18]. In short, following the 2 h euglycemic hyperinsulinemic part of the combined clamp (glucose 5 mmol/L, insulin 600 pmol/L), a 1 h rest period was included to clear the exogenously administered insulin. Thereafter, a hyperglycemic clamp was started, kee** blood glucose at 15 mmol/L for MetS and T2DM and at 10 mmol/L in controls. Blood samples were taken at fasting conditions, i.e. 5 min before the initiation of the clamp and at 70 min after the start of the glucose clamp in patients and controls. All T2DM patients were on metformin monotherapy. Research involving human subjects (including human material or human data) has been performed with the approval of our local Medical Ethical Committee (in Dutch: Medisch Ethische Toetsingscommissie or METc, of the VU Medical Center (VUmc)). The research carried out on humans is in compliance with the Helsinki Declaration. Written informed consent was obtained from each participant.
Gene expression profiling
Blood was withdrawn in PAXgene tubes (PreAnalytix, GmbH, Germany) at baseline, i.e. 5 min before the glucose clamp test, and 70 min after the hyperglycemic clamp of 10 mm/L l for controls or 15 mm/L for T2DM patients and individuals with MetS .
Total RNA was isolated from peripheral blood using the PAXgene RNA isolation kit according to the manufacturers’ instructions including a DNAse (Qiagen, Venlo, The Netherlands) step to remove genomic DNA. RNA samples were further processed by ServiceXS (Leiden, The Netherlands). Amplification was performed using the Ambion® Illumina TotalPrep RNA Amplification Kit (Ambion, # IL1791), resulting in biotinylated, amplified cRNA. Labelled RNA samples were hybridized to Sentrix Human HT12v3 Expression bead chip arrays (Illumina, San Diego, CA). Signal was developed with streptavidin-Cy3 and the BeadChip was scanned with the Illumina BeadArray Reader, which is a two-channel, 0.8 μm resolution confocal laser scanner, followed by feature extraction. Bead summary intensities were log2-transformed and normalized using quantile normalization [19].
Statistical and biological pathway analysis
We used Significance Analysis of Microarrays (SAM) [20] for the identification of over- or under-expressed genes after the glucose clamp, applying a two-class paired comparison between samples before and after the clamps. For identification of differences between study groups, we performed a two-class unpaired comparison between MetS - and T2DM individuals. Genes with a false discovery rate (FDR) < 5 % were considered significantly different. Hierarchical clustering [21] of samples was used to visualize the correlation of co-expressed genes in Treeview. For this purpose the genes were expressed relative to the median expression level in all samples (median centered data). For an interpretation of the biological processes that are represented by the genes that show a significantly different level of expression after glucose clamp or between patient groups we used Gene Ontology analysis using the PANTHER-v8.1 (Protein ANalysis THrough Evolutionary Relationships) Classification System at [22], a curated database of protein families, trees, subfamilies and functions. PANTHER uses the binomial statistics tool to compare our gene list to a reference list (NCBI: Homo sapiens genes) to determine the statistically significant over-representation of functional groups of genes. Pathway level analysis [23] of gene expression data was performed by gene set enrichment analysis (GSEA). Pathways comprise lists of genes with a connected biological background and can be used from several sources such as Kyoto Encyclopedia of Genes and Genomes (KEGG), Biocarta, and Reactome databases, user-defined pathways and previously published expression data sets as provided by the Broad institute in the Molecular Signatures database, as described in [24]. We made use of a combination of these pathways provided by the Broad Institute (C2v2 curated geneset), with a minimal geneset size of ten genes, using 1,000 gene set permutations to correct for multiple testing. This type of analysis allows the comparison of our own data with experimental expression data generated by others in an unbiased fashion. Transcription factor binding site analysis was performed on the 500 bp upstream regulatory region of the differentially expressed genes between T2DM and MetS. Lists of significantly different expressed genes between clinical samples were analyzed for enriched transcription factor binding sites by whole genome rVISTA software at [25], using the Transfac database of all transcription factor binding sites (TFBS) conserved in the human to mouse whole genome alignment of March 2006.
RNA isolation, cDNA synthesis, quantitative real-time PCR
Total RNA was isolated from peripheral blood using the PAXgene RNA isolation kit according to the manufacturers’ instructions including a DNAse (Qiagen, Venlo, The Netherlands) step to remove genomic DNA. RNA samples were concentrated by SpeedVac for 30 min. RNA purity and concentration was measured using NanoDrop ND-1,000 Spectrophotometer (Thermo Scientific, Breda, The Netherlands). cDNA was synthesized from 500 ng total RNA per sample using RevertAid™ H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s recommendations. Quantitative real-time polymerase chain reaction (PCR) was performed in an ABI PRISM 7900HT system (Applied Biosystems, Foster City, CA, USA), using primers designed by Primer Express version 2.0 (Applied Biosystems):
Briefly, in a 10 μl reaction volume, 4 μl of diluted cDNA, 5 μl SYBR Green PCR Master Mix (Applied Biosystems), and 0.5 uM of each gene-specific primers were mixed. Gene expression levels were calculated using an arbitrary standard curve and normalized to the human housekee** gene β-actin. Comparisons were performed using Student’s t-test (two-sided). Differences were considered statistically significant if probability values (P) were less than 0.05. Results are presented as mean ± standard error of the mean (SEM).
Results
Gene expression in peripheral blood cells from T2DM patients differs markedly from both MetS individuals and healthy controls
We compared the baseline gene expression levels of peripheral blood cells from six patients with T2DM to four subjects with MetS, who showed no difference in age, sex, BMI, waist circumference and blood pressure (Table 1). As expected, T2DM patients showed higher fasting plasma glucose (FPG), glycated hemoglobin A1C (HbA1c), while the lipid profile also differed, with higher triglyceride - and lower HDL levels in T2DM, compared to MetS. For comparison, we included healthy lean controls with normal fasting glucose levels and lipid profile, albeit of younger age.
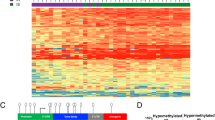
Applying statistical analysis of microarray data (SAM) [20], we identified many genes that were expressed at different levels in T2DM patients versus MetS subjects. We identified 591 genes which were expressed at higher levels and 260 genes expressed at lower levels in T2DM, compared to MetS individuals (at a false discovery rate, FDR of < 5 %, see Additional files 1 and 2). In Fig. 1 the differential transcript expression levels in circulating blood cells are visualized after hierarchical clustering of the genes. Although the lean controls were not part of this statistical analysis, we visualized their expression levels for the genes that were differentially expressed between T2DM and MetS to mark the strong resemblance of lean controls with the MetS subjects. As expected, an increased expression of proinflammatory marker genes was detected in T2DM patients, compared to MetS. These genes include: S100A12 (or EN-RAGE, extracellular newly identified receptor for advanced glycation end products-binding protein, (2.6 fold increase in T2DM, FDR = 1.97 %), CD164 (2.75 fold increase, FDR < 0.1 %), TLR1 (2.8 fold increase, FDR <0.1 %). In addition, T2DM patients expressed higher levels of the regulator of energy balance, leptin (1.5 fold increase, FDR = 4.1 %), and of the anti-oxidant gene catalase (2.0 fold increase, FDR = 4.1 %).

Visualization of 851 transcripts with significant different expression levels (FDR < 5 %) in circulating blood cells between patients with T2DM and subjects with MetS. Although the healthy lean controls were not included in the analysis, their profile is included for comparison. Blue indicates relative low expression, yellow represents a relative high expression, black represents intermediate expression
Pathway and gene set analysis identifies a reovirus signature in T2DM patients
The Panther classification system was used to determine which biological processes were represented by the differentially expressed genes in the T2DM patients versus MetS individuals by applying the statistical overrepresentation test (Fisher’s exact). Both the group of genes that were expressed at higher levels and the panel of genes which were expressed at lower levels were involved in broadly defined metabolic processes (Table 2). These data indicate that a disturbance of systemic metabolism is reflected in gene expression patterns in peripheral blood cells in patients with T2DM. Applying the same analysis in T2DM patients versus controls, we also detected significant metabolic differences, albeit at lower levels of statistical significance, due to the low number of controls.
To obtain more insight into the pathology behind the differences in gene expression, we applied pathway analysis using Gene Set Enrichment Analysis (GSEA, [23]). This analysis allowed us to compare a wide array of published genome expression data sets, defined by experimental conditions or disease states, to our own data (Table 3 and Fig. 2). Strikingly, from over 1,500 pathways (GSEA functional gene set, C2v2) analyzed, the most significant T2DM-associated gene set, comprised genes which were previously identified as being upregulated by reovirus infection [26]. Concordantly, also one of the most significant pathways for genes which were expressed at lower levels in T2DM vs MetS patients were genes downregulated by reovirus infection. The reovirus signature was generated in vitro in Human Embryonic Kidney (HEK293) cells. Because of this unexpected finding we searched for other published virus-induced genesets derived from infected circulating blood cells, thus more comparable to our study. We found two more relevant and more physiological data sets, i.e. ex vivo virus infection-induced profiles in PBMC from infected children. One set was derived from children infected with a closely related RNA virus, Rotavirus, another member of the Reoviridae family [27]. Another set was derived from a study in which infection with a more distantly related RNA virus, the Influenza virus was analyzed [28]. In both studies gene expression in PBMC from infected children was compared to PBMC from healthy children. The Rotavirus signature was also significantly associated with T2DM, Rotavirus-induced genes were upregulated in T2DM patients (Fig. 2 and Table 3), while Rotavirus-repressed genes were downregulated in T2DM patients. The signature of the Influenza virus was not associated with T2DM (Influenza-induced genes, FDR = 0.43 (or 43 %), Influenza-repressed genes, FDR = 0.14, data not shown). Because type I interferon is typically induced by viral infections we also included an ex vivo type I interferon-induced geneset in PBMC from IFNα-treated patients [29]. The type I IFN-induced geneset was also significantly increased in T2DM (Table 3 and Fig. 2). The IFN response in T2DM was also indicated by the significant geneset: GRANDVAUX_IRF3_UP, containing genes up-regulated in Jurkat T cells by expression of a constitutively active form of IRF3, which is normally activated during viral infections and induces type I IFNs [30]. In Fig. 2 the reoviral signatures are represented, showing the enriched genes: 73 reoviral genes were enriched in T2DM; expressed at high levels in T2DM out of the 227 reovirus-induced genes and also 73 reovirus-repressed genes were enriched; expressed at low levels in T2DM out of the 214 reovirus-repressed genes. Similar results are shown for the Rotavirus- and IFNα-induced signatures. Thus, gene expression profiles in peripheral blood cells induced by T2DM show a significant and specific pattern, similar to the modulatory effect of reovirus infection. We included a Venn diagram (Fig. 3), visualizing the overlap of the enriched genes in T2DM patients for the four virus infection-associated genesets. This diagram shows that the genes induced by the three virus types all contain IFNα-induced genes, in particular Influenza (11 out of 28 genes), and that 57 genes are uniquely induced by reovirus.

GSEA analysis identifies reovirus-regulated gene sets in T2DM vs MetS patients and reduced expression of cytotoxic genes in T2DM. Visualization of the enriched Reoviridae family-upregulated - and downregulated genes, IFNA-induced genes, and specific cytotoxic cell genes in T2DM patients. Blue indicates relative low expression, red represents a relative high expression

Venn diagram of enriched genes in T2DM from 4 genesets. Visualization of the overlap of the enriched genes in T2DM patients for the four virus infection-associated genesets
Although the healthy controls were not matched for age and BMI, we did perform pathway analysis and confirmed the previously reported reduced expression of genes representing electron transport chain and oxidative phosphorylation in T2DM compared to significantly younger lean controls (FDR < 0.015, data not shown) [15], while the reovirus signatures were also highly significantly present when we compared T2DM to young lean controls (data not shown).
Both T2DM and reoviral infection induce HIF-1 dependent metabolic gene profiles in peripheral blood cells
We next analyzed all genes which were upregulated by reovirus (n = 226), as published [26] and for the genes which were both upregulated by reovirus and by T2DM (n = 73), as identified by our GSEA analysis for involvement in metabolic processes. The same gene ontology analysis was performed for the genes which were downregulated by reovirus. In all of these analyses, metabolic processes were (among) the most significant processes represented by all gene sets, indicating that reovirus induces similar metabolic changes as the metabolic abnormalities observed in T2DM patients (Table 4). The 57 unique reovirus-induced genes, indicated in Fig. 3, indeed represent the metabolic processes from Table 4; nucleobase, nucleoside, nucleotide and nucleic acid metabolic process, primary metabolic processes, metabolic processes, protein amino acid phosphorylation (all p < 0.001) and monosaccharide metabolic process (p = 0.004).
To identify which factor(s) may have caused the altered gene expression in T2DM patients, we performed a transcription factor binding site analysis (Table 5). The most significant enriched binding site in the upstream regulatory region of genes which were expressed at higher levels in T2DM patients versus MetS was HIF1 (p = 1.25 × 10−12, Table 5A). HIF1 comprises the binding site for the heterodimeric protein consisting of HIF-1α and the constitutively expressed HIF-1β (also termed ARNT) and is a major regulator of metabolic processes as its activity is controlled by either hypoxia or oxidative stress. Interestingly, also ARNT binding sites were significantly overexpressed in this gene set (p = 2.33 × 10−6, Table 5A). In a comparison between T2DM patients and lean controls, the significantly higher expressed genes in T2DM also showed enriched binding sites for HIF1 and ARNT (p = 1 × 10−16 and p = 7.6 × 10−9 respectively), for lower expressed genes HIF1 was again also enriched (p = 7.1 × 10−5, data not shown). Our data thus indicate that HIF-1α/ARNT may regulate the expression of the genes involved in metabolic processes as identified from the gene ontology analysis.
Identification of a cytotoxicity profile with a blunted response to in vivo acute hyperglycemia in T2DM blood cells
To study the impact of acute hyperglycemia on the expression patterns in circulating leukocytes in the respective study groups, we analyzed alterations in gene expression profiles in T2DM patients, individuals with MetS and healthy controls in response to a hyperglycemic clamp (paired analysis, ANOVA). Only a limited number of genes per group changed in expression after exposure to acute elevations of blood glucose, and none of the genes showed altered expression upon hyperglycemia in all three groups (Table 6). The T2DM group hardly responded to the hyperglycemic clamp as only 3 genes were down-regulated (TSC22D3, DUSP1, and MCL1). In individuals with MetS, four genes were down-regulated after the 70 min hyperglycemic clamp, including C1orf162, Granzym B, FKBP5, and TSC22D3. Two genes were significantly up-regulated by high glucose levels in the MetS group: IL-8 en HIV-1 Tat specific factor one pseudogene (LOC401233). In the control group we identified nine genes which were down-regulated after in vivo exposure to high glucose, while none of the genes showed up-regulation. Remarkably, five out of nine glucose-responsive down-regulated genes in controls have a documented role in immune cell-induced cytoxicity: chloride intracellular channel 3 (CLIC3), killer cell immunoglobulin-like receptor, two domains, long cytoplasmic tail, 4 (KIR2DL4), perforin 1 (PRF1), Granzym B (GZMB) and granulysin (GNLY). These genes are typically expressed in cells with cytotoxic functions, like NK cells [55], encoding a sialomucin expressed on peripheral blood monocyte, involved in adhesion [56], and TLR1. Interestingly, mRNA encoding for the appetite hormone leptin was also increased in T2DM vs MetS, which is normally expressed in adipose tissue, and at higher levels per gram tissue from obese subjects compared to lean individuals [57, 58].
In search of transcription factors that may have caused the altered expression profile in T2DM patients, we identified highly enriched transcription factor binding sites for HIF1 and ARNT in genes with an aberrant expression compared to MetS patients (and lean controls). HIF1 is a heterodimer consisting of HIF-1α and constitutively expressed ARNT (also termed HIF-1β) and regulates the response to hypoxia and growth factors by controlling energy metabolism [59]. Indeed, gene ontology analysis confirmed that metabolic processes were affected in T2DM patients compared to circulating cells from MetS subjects. Affected metabolic processes have been demonstrated before in blood cells from T2DM versus MetS, or versus nondiabetic controls [60, 61], although the subjects were not matched for BMI as in our study. It may not be surprising to identify metabolic processes in metabolically disturbed patients, however it does indicate that these metabolic changes are reflected in the gene expression profile of peripheral blood cells and can thus be monitored. Interestingly, HIF-1α activity, experimentally induced upon intermittent hypoxia, increases plasma triglycerides through upregulation of sterol regulatory element binding protein (SREBP)-1 activity in the liver [62]. These results correspond to our findings that T2DM patients show higher HIF-1α activity, associated with higher plasma triglyceride levels. Furthermore, the genes showing higher transcript levels in T2DM patients not only contained binding sites for HIF-1α and ARNT, but also for SREBP-1, thus reminiscent of the molecular switches during hypoxia in the liver.
The previously described reduced expression of genes involved in electron transport chain and oxidative phosphorylation in PBMC from T2DM vs young lean controls [15] is consistent with increased HIF-1α activity in T2DM. We confirmed the reduced expression of electron transport chain and oxidative phosphorylation genes in T2DM when we compared these patients to young lean controls (Additional file 1B). It is quite remarkable that the analysis of only three controls and six T2DM patients results in such highly significant pathways which are consistent with previously published data.
It is not quite clear what exactly caused the increased activity of HIF-1α in T2DM patients. In normal physiology, hypoxia regulates HIF-1α activity on the protein level by preventing its degradation by an oxygen-dependent mechanism, reviewed in [63]. However, it has been suggested before that hyperglycemia mimics the effects of hypoxia, in terms of an increased cytosolic ratio of NADH/NAD+, referred to as pseudo hypoxia in diabetes patients [64]. Reactive oxygen species (ROS) may also stabilize HIF-1α under normoxic conditions, resulting in increased glycolytic metabolism and increased NADH/NAD+ ratio [65]. High dietary fat intake and obesity causes increased release of skeletal muscle mitochondrial ROS species (H2O2) accompanied by a change in redox environment to a more oxidized state (generally referred to as oxidative stress) and insulin resistance in mice and humans [66] Targeting the altered redox state in mice by neutralization of H2O2 by an antioxidant or by overexpression of catalase in mitochondria of muscle cells prevents high fat diet-induced insulin resistance [66]. The two-fold increased expression of catalase in circulating cells from T2DM patients compared to MetS suggests that a compensatory mechanism for increased oxidative damage is activated in T2DM. Therefore, it might very well be that in T2DM patients, chronic hyperglycemia and oxidative stress contributes to the HIF-1α- regulated metabolic change in peripheral blood cells. Indeed, prolonged high glucose levels in vivo induces the expression of the HIF-1α-responsive gene VEGF and vascular dysfunction in rats [67]. Interestingly, adipocyte-specific disruption of ARNT or disruption of HIF-1α in mice results in similar metabolic phenotypes. In both mouse models the high fat diet-induced abnormalities are reduced, including diminished fat formation, protection from obesity and insulin resistance [68], suggesting a role for HIF1 in the pathogenesis of obesity and insulin resistance.
The gene expression profile in peripheral blood seems to partially mimic the profile of adipocytes, with respect to HIF-1α activity and leptin expression (both associated with obesity), and to partially mimic the response in the liver, reflected by the enriched binding sites for SREBP-1 normally activated by HIF-1α in the liver. These findings are in line with the suggestion that altered gene expression in specific organs, is reflected in peripheral blood cells [14].
Conclusions
Using an unbiased approach we have revealed that a hyperglycemic state affects the cytotoxic immune capacity. The reduced cytotoxic potential may render the T2DM patients less capable to clear viral infections. This suggestion is supported by the identification of a reovirus expression signature in T2DM patients. It is tempting to speculate that viral infections in T2DM fuel systemic inflammatory processes and may even lead to infection of pancreatic beta cells, as has been shown in experimental animals, and thus contribute to the induction of insulitis and insulin-dependent diabetes.
References
Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–5.
Laakso M. Cardiovascular disease in type 2 diabetes from population to man to mechanisms: the Kelly West Award Lecture 2008. Diabetes Care. 2010;33:442–9.
Turnbull FM, Abraira C, Anderson RJ, Byington RP, Chalmers JP, Duckworth WC, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. 2009;52:2288–98.
Standl E. Statins and beyond: concurrent strategies for prevention of cardiovascular disease in patients with type 2 diabetes. Diab Vasc Dis Res. 2013;10:99–114.
Coutinho M, Gerstein HC, Wang Y, Yusuf S. The relationship between glucose and incident cardiovascular events. A metaregression analysis of published data from 20 studies of 95,783 individuals followed for 12.4 years. Diabetes Care. 1999;22:233–40.
DECODE Study Group tEDEG. Glucose tolerance and cardiovascular mortality: comparison of fasting and 2-h diagnostic criteria. Arch Intern Med. 2001;161:397–405.
Donath MY, Shoelson SE. Type 2 diabetes as an inflammatory disease. Nat Rev Immunol. 2011;11:98–107.
Richardson VR, Smith KA, Carter AM. Adipose tissue inflammation: feeding the development of type 2 diabetes mellitus. Immunobiology. 2013;218:1497–504.
Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation–mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol. 2012;32:1771–6.
Donath MY, Schumann DM, Faulenbach M, Ellingsgaard H, Perren A, Ehses JA. Islet inflammation in type 2 diabetes: from metabolic stress to therapy. Diabetes Care. 2008;31 Suppl 2:S161–4.
Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, et al. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–70.
Eguchi K, Manabe I. Macrophages and islet inflammation in type 2 diabetes. Diabetes Obes Metab. 2013;15 Suppl 3:152–8.
Kempf K, Rose B, Herder C, Haastert B, Fusbahn-Laufenburg A, Reifferscheid A, et al. The metabolic syndrome sensitizes leukocytes for glucose-induced immune gene expression. J Mol Med (Berl). 2007;85:389–96.
Liew CC, Ma J, Tang HC, Zheng R, Dempsey AA. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J Lab Clin Med. 2006;147:126–32.
Takamura T, Honda M, Sakai Y, Ando H, Shimizu A, Ota T, et al. Gene expression profiles in peripheral blood mononuclear cells reflect the pathophysiology of type 2 diabetes. Biochem Biophys Res Commun. 2007;361:379–84.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Alberti KG, Zimmet P, Shaw J. Metabolic syndrome–a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23:469–80.
Bunck MC, Diamant M, Corner A, Eliasson B, Malloy JL, Shaginian RM, et al. One-year treatment with exenatide improves beta-cell function, compared with insulin glargine, in metformin-treated type 2 diabetic patients: a randomized, controlled trial. Diabetes Care. 2009;32:762–8.
Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93.
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21.
Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8.
Mi H, Muruganujan A, Thomas PD. PANTHER in 2013: modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013;41:D377–86.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50.
van der Pouw Kraan TC, van der Laan AM, Piek JJ, Horrevoets AJ. Surfing the data tsunami, a bioinformatic dissection of the proangiogenic monocyte. Vascul Pharmacol. 2012;56:297–305.
Zambon AC, Zhang L, Minovitsky S, Kanter JR, Prabhakar S, Salomonis N, et al. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc Natl Acad Sci U S A. 2005;102:8561–6.
DeBiasi RL, Clarke P, Meintzer S, Jotte R, Kleinschmidt-Demasters BK, Johnson GL, et al. Reovirus-induced alteration in expression of apoptosis and DNA repair genes with potential roles in viral pathogenesis. J Virol. 2003;77:8934–47.
Wang Y, Dennehy PH, Keyserling HL, Tang K, Gentsch JR, Glass RI, et al. Rotavirus infection alters peripheral T-cell homeostasis in children with acute diarrhea. J Virol. 2007;81:3904–12.
Ramilo O, Allman W, Chung W, Mejias A, Ardura M, Glaser C, et al. Gene expression patterns in blood leukocytes discriminate patients with acute infections. Blood. 2007;109:2066–77.
Ji X, Cheung R, Cooper S, Li Q, Greenberg HB, He XS. Interferon alfa regulated gene expression in patients initiating interferon treatment for chronic hepatitis C. Hepatology. 2003;37:610–21.
Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, et al. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol. 2002;76:5532–9.
Dybkaer K, Iqbal J, Zhou G, Geng H, **ao L, Schmitz A, et al. Genome wide transcriptional analysis of resting and IL2 activated human natural killer cells: gene expression signatures indicative of novel molecular signaling pathways. BMC Genomics. 2007;8:230–47.
Obata-Onai A, Hashimoto S, Onai N, Kurachi M, Nagai S, Shizuno K, et al. Comprehensive gene expression analysis of human NK cells and CD8(+) T lymphocytes. Int Immunol. 2002;14:1085–98.
Correia MP, Costa AV, Uhrberg M, Cardoso EM, Arosa FA. IL-15 induces CD8+ T cells to acquire functional NK receptors capable of modulating cytotoxicity and cytokine secretion. Immunobiology. 2011;216:604–12.
Palmer C, Diehn M, Alizadeh AA, Brown PO. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics. 2006;7:115.
Bouma G, Baggen JM, van Bodegraven AA, Mulder CJ, Kraal G, Zwiers A, et al. Thiopurine treatment in patients with Crohn’s disease leads to a selective reduction of an effector cytotoxic gene expression signature revealed by whole-genome expression profiling. Mol Immunol. 2013;54:472–81.
Bloom ET, Umehara H, Bleackley RC, Okumura K, Mostowski H, Babbitt JT. Age-related decrement in cytotoxic T lymphocyte (CTL) activity is associated with decreased levels of mRNA encoded by two CTL-associated serine esterase genes and the perforin gene in mice. Eur J Immunol. 1990;20:2309–16.
Khan N, Hislop A, Gudgeon N, Cobbold M, Khanna R, Nayak L, et al. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J Immunol. 2004;173:7481–9.
Dimayuga PC, Chyu KY, Lio WM, Zhao X, Yano J, Zhou J, et al. Reduced neointima formation after arterial injury in CD4−/− mice is mediated by CD8 + CD28hi T cells. J Am Heart Assoc. 2013;2, e000155.
Chyu KY, Zhao X, Dimayuga PC, Zhou J, Li X, Yano J, et al. CD8+ T cells mediate the athero-protective effect of immunization with an ApoB-100 peptide. PLoS One. 2012;7, e30780.
Smyth MJ, Kelly JM, Sutton VR, Davis JE, Browne KA, Sayers TJ, et al. Unlocking the secrets of cytotoxic granule proteins. J Leukoc Biol. 2001;70:18–29.
Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91.
Nicolucci A. Epidemiological aspects of neoplasms in diabetes. Acta Diabetol. 2010;47:87–95.
Lin TM, Chen WJ, Chen HY, Wang PW, Eng HL. Increased incidence of cytomegalovirus but not Chlamydia pneumoniae in atherosclerotic lesions of arteries of lower extremities from patients with diabetes mellitus undergoing amputation. J Clin Pathol. 2003;56:429–32.
Diepersloot RJ, Bouter KP, Beyer WE, Hoekstra JB, Masurel N. Humoral immune response and delayed type hypersensitivity to influenza vaccine in patients with diabetes mellitus. Diabetologia. 1987;30:397–401.
Diepersloot RJ, Bouter KP, Van BR, Lucas CJ, Masurel N, Erkelens DW. Cytotoxic T-cell response to influenza A subunit vaccine in patients with type 1 diabetes mellitus. Neth J Med. 1989;35:68–75.
Onodera T, Jenson AB, Yoon JW, Notkins AL. Virus-induced diabetes mellitus: reovirus infection of pancreatic beta cells in mice. Science. 1978;201:529–31.
Jun HS, Yoon JW. A new look at viruses in type 1 diabetes. Diabetes Metab Res Rev. 2003;19:8–31.
Graham KL, Sanders N, Tan Y, Allison J, Kay TW, Coulson BS. Rotavirus infection accelerates type 1 diabetes in mice with established insulitis. J Virol. 2008;82:6139–49.
Honeyman MC, Laine D, Zhan Y, Londrigan S, Kirkwood C, Harrison LC. Rotavirus infection induces transient pancreatic involution and hyperglycemia in weanling mice. PLoS One. 2014;9, e106560.
Yeung WC, Rawlinson WD, Craig ME. Enterovirus infection and type 1 diabetes mellitus: systematic review and meta-analysis of observational molecular studies. BMJ. 2011;342:d35.
Ylipaasto P, Smura T, Gopalacharyulu P, Paananen A, Seppanen-Laakso T, Kaijalainen S, et al. Enterovirus-induced gene expression profile is critical for human pancreatic islet destruction. Diabetologia. 2012;55:3273–83.
Richardson SJ, Willcox A, Bone AJ, Foulis AK, Morgan NG. The prevalence of enteroviral capsid protein vp1 immunostaining in pancreatic islets in human type 1 diabetes. Diabetologia. 2009;52:1143–51.
van Asseldonk EJ, Stienstra R, Koenen TB, Joosten LA, Netea MG, Tack CJ. Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab. 2011;96:2119–26.
Kosaki A, Hasegawa T, Kimura T, Iida K, Hitomi J, Matsubara H, et al. Increased plasma S100A12 (EN-RAGE) levels in patients with type 2 diabetes. J Clin Endocrinol Metab. 2004;89:5423–8.
Tang J, Luo Z, Zhou G, Song C, Yu F, ** HIF-1alpha/E-box/AP-1-like sequences of CD164. BMC Mol Biol. 2011;12:44–54.
Watt SM, Buhring HJ, Rappold I, Chan JY, Lee-Prudhoe J, Jones T, et al. CD164, a novel sialomucin on CD34(+) and erythroid subsets, is located on human chromosome 6q21. Blood. 1998;92:849–66.
Fried SK, Ricci MR, Russell CD, Laferrere B. Regulation of leptin production in humans. J Nutr. 2000;130:3127S–31.
Galic S, Oakhill JS, Steinberg GR. Adipose tissue as an endocrine organ. Mol Cell Endocrinol. 2010;316:129–39.
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–32.
Manoel-Caetano FS, Xavier DJ, Evangelista AF, Takahashi P, Collares CV, Puthier D, et al. Gene expression profiles displayed by peripheral blood mononuclear cells from patients with type 2 diabetes mellitus focusing on biological processes implicated on the pathogenesis of the disease. Gene. 2012;511:151–60.
Grayson BL, Wang L, Aune TM. Peripheral blood gene expression profiles in metabolic syndrome, coronary artery disease and type 2 diabetes. Genes Immun. 2011;12:341–51.
Li J, Bosch-Marce M, Nanayakkara A, Savransky V, Fried SK, Semenza GL, et al. Altered metabolic responses to intermittent hypoxia in mice with partial deficiency of hypoxia-inducible factor-1alpha. Physiol Genomics. 2006;25:450–7.
Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol. 2010;345:105–20.
Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42:801–13.
Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome-a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–52.
Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J Clin Invest. 2009;119:573–81.
Tilton RG, Kawamura T, Chang KC, Ido Y, Bjercke RJ, Stephan CC, et al. Vascular dysfunction induced by elevated glucose levels in rats is mediated by vascular endothelial growth factor. J Clin Invest. 1997;99:2192–202.
Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, et al. Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes. 2011;60:2484–95.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CTMPK participated in the design of the study, analyzed the data and drafted the manuscript. WJC, MCMB, DHR, NJZ, REG, LB performed the hyperglycemic clamps, participated in coordination of the study and helped to draft the manuscript. JMB processed blood samples and performed gene expression quantification. EHS partly coordinated the study, and helped to draft the manuscript. MD and AJGH designed and supervised the study, and contributed to drafting the manuscript. All authors read and approved the final manuscript.
Michaela Diamant deceased.
Additional files
Additional file 1:
Genes expressed at higher levels in T2DM patients compared to individuals with MetS. Statistical analysis was performed using the SAM package. A q value <5 % is considered significant.
Additional file 2:
Genes expressed at lower levels in T2DM patients compared to individuals with MetS. Statistical analysis was performed using the SAM package. A q value <5 % is considered significant.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
van der Pouw Kraan, T.C.T.M., Chen, W.J., Bunck, M.C.M. et al. Metabolic changes in type 2 diabetes are reflected in peripheral blood cells, revealing aberrant cytotoxicity, a viral signature, and hypoxia inducible factor activity. BMC Med Genomics 8, 20 (2015). https://doi.org/10.1186/s12920-015-0096-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-015-0096-y