
Abstract
Background
Seneca Valley virus (SVV) has emerged in multiple countries in recent years. SVV infection can cause vesicular lesions clinically indistinguishable from those caused by other vesicular disease viruses, such as foot-and-mouth disease virus (FMDV), swine vesicular disease virus (SVDV), vesicular stomatitis virus (VSV), and vesicular exanthema of swine virus (VESV). Sensitive and specific RT-PCR assays for the SVV detection is necessary for differential diagnosis. Real-time RT-PCR (rRT-PCR) has been used for the detection of many RNA viruses. The insulated isothermal PCR (iiPCR) on a portable POCKIT™ device is user friendly for on-site pathogen detection. In the present study, SVV rRT-PCR and RT-iiPCR were developed and validated.
Results
Neither the SVV rRT-PCR nor the RT-iiPCR cross-reacted with any of the vesicular disease viruses (20 FMDV, two SVDV, six VSV, and two VESV strains), classical swine fever virus (four strains), and 15 other common swine viruses. Analytical sensitivities of the SVV rRT-PCR and RT-iiPCR were determined using serial dilutions of in vitro transcribed RNA as well as viral RNA extracted from a historical SVV isolate and a contemporary SVV isolate. Diagnostic performances were further evaluated using 125 swine samples by two approaches. First, nucleic acids were extracted from the 125 samples using the MagMAX™ kit and then tested by both RT-PCR methods. One sample was negative by the rRT-PCR but positive by the RT-iiPCR, resulting in a 99.20% agreement (124/125; 95% CI: 96.59–100%, κ = 0.98). Second, the 125 samples were tested by the taco™ mini extraction/RT-iiPCR and by the MagMAX™ extraction/rRT-PCR system in parallel. Two samples were positive by the MagMAX™/rRT-PCR system but negative by the taco™ mini/RT-iiPCR system, resulting in a 98.40% agreement (123/125; 95% CI: 95.39–100%, κ = 0.97). The two samples with discrepant results had relatively high CT values.
Conclusions
The SVV rRT-PCR and RT-iiPCR developed in this study are very sensitive and specific and have comparable diagnostic performances for SVV RNA detection. The SVV rRT-PCR can be adopted for SVV detection in laboratories. The SVV RT-iiPCR in a simple field-deployable system could serve as a tool to help diagnose vesicular diseases in swine at points of need.
Similar content being viewed by others


Background
Seneca Valley virus (SVV) is a single-stranded, positive-sense RNA virus belonging to the species Senecavirus A in the genus Senecavirus in the family Picornaviridae [1, 2]. Although the species name Senecavirus A has been used in some publications as the virus name with an acronym of SVA, in fact the virus name is Seneca Valley virus. The SVV genome (approximately 7.3 kb) contains a single open reading frame (ORF) flanked by a long 5′ untranslated region (UTR; 668 nucleotides) and a short 3′ UTR (68 nucleotides) followed by a poly(A) tail. The polyprotein translated from the single ORF is predicted to be post-translationally processed into four structural proteins (VP4, VP2, VP3, and VP1) and seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) [2].
SVV was initially incidentally identified as a contaminant in PER.C6 cell cultures in 2002 [2]. From 1988 to 2001, a number of virus isolates were sporadically recovered from pigs in various U.S. states but with no detailed description of the clinical symptoms [3]. Sequence analysis of these retrospective virus isolates suggested that these viruses were the same as SVV. Thereafter, SVV was sporadically identified in pigs with swine idiopathic vesicular disease in Canada in 2007 [4] and in the U.S. in 2010 [5], but not much attention was drawn to this virus. At the end of 2014 and the beginning of 2015, multiple outbreaks of vesicular disease in weaned and adult pigs as well as increasing mortality rates of neonatal piglets (1–4 days of age) were reported in Brazil [6,7,8]. SVV was consistently detected from the pigs with vesicular lesions while other vesicular viral pathogens were not detected [9]. Starting from July 2015, SVV was consistently detected from increasing swine idiopathic vesicular disease cases observed in exhibition, commercial finisher, and breeding swine herds in the U.S. [10]. Foreign animal disease investigations indicated that other vesicular viral pathogens, such as foot-and-mouth disease virus (FMDV), swine vesicular disease virus (SVDV), vesicular stomatitis virus (VSV), and vesicular exanthema of swine virus (VESV), were negative in these cases [10]. Subsequently, SVV detection was reported by other laboratories in the U.S. [11,12,13,14,15,16], China [17,18,19,20,21], Canada [22], Thailand [23], and Colombia [24]. Vesicular lesions were induced in pigs following experimental inoculation with the contemporary U.S. isolates of SVV [25, 26], confirming that SVV is a vesicular viral pathogen. In one experimental infection study [27], a historical SVV isolate (SVV-001) did not cause overt clinical diseases in the inoculated pigs but it established infection in pigs and induced an immune response. Since the vesicular lesions caused by SVV infection are clinically indistinguishable from those caused by other vesicular disease viruses (e.g., FMDV, SVDV, VSV, and VESV), differential diagnosis is mandatory. RT-PCR is a sensitive and fast method commonly used to differentiate vesicular viral pathogens.
A number of SVV specific gel-based (conventional) RT-PCR, nested RT-PCR, real-time RT-PCR (rRT-PCR), reverse transcription droplet digital PCR, and loop-mediated isothermal amplification assays have been described in the literatures although not all of them have been fully validated [6, 28,29,30,31,32,33,34,35]. Compared to the conventional RT-PCR assays, rRT-PCR is generally more sensitive and suitable for high throughput testing with shorter turnaround time. It is noteworthy that conduction of rRT-PCR assays requires trained technicians and expensive instruments; rRT-PCR assays are mainly performed in the laboratory rather than for on-site applications.
In recent years, a fluorescent hydrolysis probe-based insulated isothermal PCR (iiPCR) technology has been described [36]. The iiPCR and RT-iiPCR can be used for the detection of DNA and RNA molecules. The principle of the iiPCR is to amplify the DNA/RNA by cycling the reaction components through different temperature gradients (denaturation, annealing, and extension) in a capillary tube on a simple Nucleic Acid Analyzer [36, 37]. The iiPCR technology and a commercially available field-deployable device (POCKIT™ combo system), which includes a taco™ mini Automatic Nucleic Acid Extraction System and a POCKIT™ Nucleic Acid Analyzer (GeneReach USA, Lexington, MA, USA), allow automatic detection and interpretation of PCR results within 1–1.5 h [36]. It has been shown that iiPCR or RT-iiPCR assays have excellent sensitivity and specificity for the detection of various targets, including swine pathogens, such as classical swine fever virus (CSFV), FMDV, porcine epidemic diarrhea virus (PEDV), porcine deltacoronavirus (PDCoV), and porcine reproductive and respiratory syndrome virus (PRRSV) [38,39,40,41], and various pathogens in shrimp, dogs, cats, poultry, ruminants, and horses [42,43,44,45,46,47,48,49,50,51,52].
In the present study, a SVV rRT-PCR targeting the conserved 5′ UTR and a SVV RT-iiPCR targeting the conserved 3D gene region were developed and validated for the detection of SVV RNA.
Results
Genetic diversity of SVV and design of the primers and probes for the SVV real-time RT-PCR and SVV RT-iiPCR
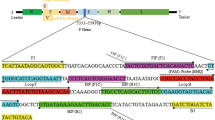
The VP1 sequences are generally used to assess the genetic diversity of SVV. Phylogenetic analyses of the VP1 nucleotide sequences indicated that the global SVV strains formed into the clade of historical SVV (detected in the USA from 1988 to 2001) and the clade of contemporary SVV (detected in the USA, Brazil, Canada, China, Colombia, and Thailand mainly since 2015) (Fig. 1). Based on the VP1 sequences, 112 global SVV strains had 86.2–100% nucleotide identities, with 93.3–99.9% nucleotide identities among the historical clade, 94.2–100% among the contemporary clade, and 86.2–94.8% between the historical and contemporary clades. The 79 SVV strains with whole genome sequences available can well represent the genetic diversity of SVVs (Fig. 1) and these whole genome sequences were used to design the primers and probes targeting the conserved genomic regions. The primers and probe of a SVV rRT-PCR targeting the conserved 5′ UTR and the primers and probe of a SVV RT-iiPCR targeting the 3D gene are shown in Table 1.

Phylogenetic analysis of global SVV strains. The VP1 sequences of 112 global SVV strains available in GenBank were analyzed to assess the genetic diversity of SVVs. The 79 SVV strains with the whole genome sequences available are denoted with a black triangle. SVV strains from different countries are denoted with different colors. The GenBank accession numbers are included in each strain name
Analytical specificity of SVV rRT-PCR and RT-iiPCR
As shown in Table 2, the SVV rRT-PCR and RT-iiPCR only specifically reacted with SVV and did not cross-react with any of the vesicular disease viruses that included 20 FMDV strains, two SVDV strains, six VSV strains, and two VESV strains. The SVV rRT-PCR and RT-iiPCR also did not cross-react with the four strains of CSFV and any of 15 other common swine viruses.
Analytical sensitivity of SVV rRT-PCR and RT-iiPCR
The analytical sensitivities of the SVV rRT-PCR and RT-iiPCR were determined by testing serial dilutions of in vitro transcribed (IVT) RNA. For the SVV rRT-PCR, each dilution of the SVV IVT RNA containing the 5′ UTR (108, 107, 106, 105, 104, 103, 102, 10, 1, 0.1 and 0.01 copies per μL) was run in triplicates. The standard curve had an r2 = 0.996 and a slope of − 3.01. The LoD of the rRT-PCR for SVV detection was about 3.5 RNA copies per reaction. For the SVV RT-iiPCR, testing serial dilutions of the IVT RNA containing a fragment of the 3D gene (100, 50, 20, 10, 5, and 0 copies per reaction) revealed that 5/5 (100%), 15/15 (100%), 20/20 (100%), 20/20 (100%), 15/20 (75%), and 0% (0/20) produced positive results on these RNA copies, respectively. The LoD95% of the SVV RT-iiPCR was estimated to be 7 RNA copies per reaction.
The analytical sensitivities of the SVV rRT-PCR and RT-iiPCR were also evaluated by testing RNA extracts from 10-fold serial dilutions (triplicate for each dilution) of SVV cell culture isolates (a US historical isolate SVV-001 and a US contemporary isolate USA/SD41901/2015). The 100% detection endpoints of the SVV rRT-PCR to detect the two SVV isolates were both at 10− 7 dilutions (Table 3; 3 TCID50 (50% tissue culture infectious dose)/ml). The 100% detection endpoints of the SVV RT-iiPCR was at 10− 6 dilutions for the two SVV isolates (Table 3; 30 TCID50/ml).
Performance of SVV rRT-PCR and RT-iiPCR in detecting SVV in clinical samples
The performances of the SVV rRT-PCR and RT-iiPCR for the detection of SVV RNA in swine clinical samples were evaluated by testing 125 clinical samples including 12 vesicular swabs, 30 tonsil swabs, 25 oral fluids, 28 sera and 30 fecal swabs. Distributions of the 125 clinical samples based on specimen types and CT ranges are summarized in Table 4.
Nucleic acids of the 125 clinical samples were first extracted using a MagMAX™ Pathogen RNA/DNA Kit and then the same nucleic acids were tested by SVV rRT-PCR and RT-iiPCR in parallel with results shown in Table 5. Among the 125 samples, 73 were positive and 52 were negative by SVV rRT-PCR; 74 were positive and 51 were negative by SVV RT-iiPCR. The 73 SVV rRT-PCR-positivie samples were also positive by the RT-iiPCR, while 51 of the 52 rRT-PCR-negative samples were negative by RT-iiPCR. The agreement between the two RT-PCR methods was 99.20% (95% CI: 96.59–101.81%; kappa value = 0.98). Compared to the SVV rRT-PCR, the sensitivity and specificity of the SVV RT-iiPCR were 100 and 98.08%, respectively. The one discrepant sample was retested in triplicate: 1/3 were positive by the rRT-PCR (CT = 33.1) and 2/3 were positive by the RT-iiPCR.
In the POCKIT™ Combo system, the field-deployable taco™ mini Automatic Nucleic Acid Extraction System (taco™ mini, GeneReach USA, Lexington, MA, USA) allows automatic nucleic acid extraction on site for PCR detection on the POCKIT™ Nucleic Acid Analyzer device. In order to compare the performance of the SVV RT-iiPCR in the POCKIT™ combo system to the SVV rRT-PCR conducted in the laboratory, the following approaches were used. First, nucleic acids were extracted from the 125 clinical samples using the taco™ mini automatic extraction instrument followed by RT-iiPCR testing in the POCKIT™ device (taco™ mini/RT-iiPCR). Meanwhile, nucleic acids were extracted from the same 125 samples using a MagMAX™ Pathogen RNA/DNA Kit followed by the SVV rRT-PCR on the ABI 7500 Fast instrument (MagMAX™/rRT-PCR). As shown in Table 6, among the 125 samples, 73 were positive and 52 were negative by the MagMAX™/rRT-PCR system; 71 were positive and 54 were negative by the taco™ mini/RT-iiPCR system. The 52 MagMAX™/rRT-PCR-negative samples were all negative by the SVV taco™ mini/RT-iiPCR system while two samples positive by the MagMAX™/rRT-PCR system were negative by the taco™ mini/RT-iiPCR system. The agreement between the two systems was 98.4% (95% CI: 95.39–100%; kappa value = 0.97). The two discrepant samples were further tested in triplicate: both samples were positive (3/3) by the MagMAX™/rRT-PCR system (CT = 32.2, 35.7, and 36.0 for one sample; CT = 36.2, 36.6, and 37.9 for another sample), while 0/3 and 1/3 positive by the taco™ mini/RT-iiPCR system, respectively.
Discussion
Recent emergence of SVV infection in multiple countries [6, 10, 11, 17, 23, 24] and the experimental confirmation of SVV as a vesicular viral pathogen [25,26,27] have raised the concern of differentially diagnosing vesicular diseases in pigs, because the vesicular lesions caused by SVV infection are clinically indistinguishable from those caused by other vesicular disease viruses, such as FMDV, SVDV, VSV, and VESV. The USA is currently free of some foreign animal diseases such as FMDV. Introduction of FMDV would devastate the US pork section. Thus, early detection and recognition of FMDV is critical to minimize the virus spread and economic burden. However, the increased incidence of SVV in the USA may increase the likelihood that veterinarians will assume that the presence of vesicular lesions are due to SVV and not report them to State or Federal animal health officials; this would put the early recognition of FMDV at risk. Therefore, it is critically important to conduct differential diagnosis when vesicular lesions are observed in pigs. RT-PCR is a sensitive and fast method commonly used to differentiate vesicular viral pathogens.
In the current study, we developed and evaluated a SVV rRT-PCR and a field-deployable SVV RT-iiPCR. Both RT-PCR methods are very specific and do not cross-react with other vesicular viral pathogens and other common swine viral pathogens. Both RT-PCR methods can detect the historical and contemporary SVV strains but the SVV rRT-PCR was about 10-fold more sensitive for endpoint detection compared to the SVV RT-iiPCR. The endpoint dilutions negative by the SVV RT-iiPCR but positive by the SVV rRT-PCR had high CT values (34.3–37.3). The SVV rRT-PCR and SVV RT-iiPCR had similar analytical sensitivity in terms of detection in the unit of genomic copies (LoD of 3.5 copies/reaction for SVV rRT-PCR and 7 copies/reaction for SVV RT-iiPCR). Overall, the SVV rRT-PCR and SVV RT-iiPCR had comparable analytical sensitivities.
Subsequently, the diagnostic performances of two SVV RT-PCR assays on clinical samples were evaluated. When the nucleic acids extracted from 125 clinical samples using the MagMAX™ Pathogen RNA/DNA Kit on a Kingfisher Flex instrument were tested, there was only one sample with discrepant results between the SVV rRT-PCR and RT-iiPCR (Table 5). This sample was retested in three replicates, it was found that 1/3 was positive by the rRT-PCR (CT = 33.1) and 2/3 were positive by RT-iiPCR. If the retest results were taken into account, the SVV rRT-PCR and RT-iiPCR would have had zero discrepant results. These data demonstrate excellent agreements between these two SVV RT-PCR methods.
However, for on-site detection, it is impractical to extract nucleic acids from samples using the MagMAX™ Pathogen RNA/DNA Kit on a Kingfisher Flex instrument. Since a portable POCKIT™ Combo package is commercially available that allows on-site nucleic acid extraction using the included taco™ mini instrument and on-site PCR detection using a POCKIT™ Nucleic Acid Analyzer, we further compared the clinical performances of the taco™ mini extraction/RT-iiPCR system to the MagMAX™ extraction/rRT-PCR system based on testing 125 clinical samples. Overall, 98.4% agreement was observed for the two SVV RT-PCR systems (Table 6). When the two discrepant samples were retested in triplicate, three replicates of both samples were positive (3/3) by the MagMAX™/rRT-PCR system (CT = 32.2, 35.7, and 36.0 for one sample; CT = 36.2, 36.6, and 37.9 for another sample), while 0/3 and 1/3 replicates were positive by the taco™ mini/RT-iiPCR system, respectively. Again, these two samples with discrepant results by the two SVV RT-PCR systems had relatively high CT values (low concentrations of virus); sequencing was attempted on these two samples to confirm the RT-PCR results but was unsuccessful. Overall, the data suggest that the SVV MagMAX™/rRT-PCR system and the taco™ mini/RT-iiPCR system have comparable performances on detecting SVV from clinical samples.
In the past a few years, multiple assays have been developed for rapid detection of SVV from clinical samples. These include a conventional two-step RT-PCR assay targeting the VP3/VP1 region [6], a conventional nested-PCR assay targeting the VP1 region [28], a SYBR Green-based real-time RT-PCR assay targeting the VP1 region [29], two TaqMan probe-based real-time RT-PCR assays respectively targeting the 3D region [30] and the VP1 region [31], two reverse transcription droplet digital PCR assays both targeting the 3D region [32, 33], and several reverse transcription loop-mediated isothermal amplification (RT-LAMP) assays targeting the VP1, VP2, 5′ UTR, or VP3/VP1 region [34, 35]. We did not perform a head-to-head comparison of our SVV rRT-PCR and RT-iiPCR to these previously published assays. It is hard to draw a clear conclusion on the analytical sensitivity, diagnostic performance, and cost of all of these described SVV detection assays.
Currently, the samples from pigs with suspect vesicular diseases are submitted to veterinary diagnostic laboratories or the National Veterinary Service Laboratory Foreign Animal Disease Diagnostic Laboratory for differential diagnosis. The sensitive and specific SVV rRT-PCR assay developed in this study can be adopted for SVV detection in laboratories. For the long run, it will be ideal to have some on-site tests capable of quickly and reliably differentiating vesicular viral pathogens so that prompt responses can be taken accordingly without transporting samples, which can be another risk of spreading the virus, to laboratories and waiting for the results. The sensitive and specific SVV RT-iiPCR in a simple field-deployable system described in this study could serve as a tool to help on-site diagnosis of vesicular diseases in swine.
Conclusions
An rRT-PCR and a field deployable RT-iiPCR were developed and validated for the detection of SVV in this study. Both RT-PCR methods are very specific and do not cross-react with other vesicular viral pathogens and other common swine viral pathogens. Both RT-PCR methods are very sensitive and can detect both the historical and contemporary SVV strains. Furthermore, both RT-PCR systems had comparable diagnostic performances for SVV RNA detection from clinical samples. The SVV rRT-PCR system can be adopted for SVV detection in laboratories. The SVV RT-iiPCR in a simple field-deployable system could serve as a tool to help diagnose vesicular diseases in swine at points of need.
Methods
Note: Real-time RT-PCR and RT-iiPCR have been previously described for other pathogens. Some procedures for SVV real-time RT-PCR and SVV RT-iiPCR described below are somewhat similar to the methodology previously described for PEDV and PDCoV real-time RT-PCR and RT-iiPCR [40].
Design of primers and probes for the SVV real-time RT-PCR and SVV RT-iiPCR
The VP1 sequences of 112 global SVV strains available in GenBank were used for phylogenetic analyses to assess the genetic diversity of SVVs. Alignment was conducted using the MUSCLE program and phylogenetic tree was constructed using the maximum-likelihood method of MEGA6 [53] with a bootstrap analysis of 1000 replicates (Fig. 1). The GenBank accession numbers are included in each strain name as shown in Fig. 1. Among the 112 global SVV strains, whole genome sequences were available for 79 SVV strains and they well represented the genetic diversity of SVVs (Fig. 1). Subsequently, the 79 SVV whole genome sequences were aligned and the primers and probes were designed to target the conserved genomic regions using the Primer Express software 3.0.1 (Thermo Fisher Scientific). Specifically, a SVV rRT-PCR targeting the conserved 5′ UTR and a SVV RT-iiPCR targeting the 3D gene were developed in this study (Table 1).
Viruses
A U.S. historical SVV isolate (SVV 001, ATCC-PTA-5342) obtained from the American Type Culture Collection (Manassas, VA, USA) and a U.S. contemporary SVV isolate USA/SD41901/2015 obtained at the Iowa State University Veterinary Diagnostic Laboratory (ISU VDL) [10] were used for evaluating the analytical sensitivities of the SVV RT-PCRs. Both SVV isolates were propagated and titrated in H1299 cell line obtained from the American Type Culture Collection (ATCC CRL-5803). For analytical sensitivity analysis, the two SVV cell culture isolates with infectious titers of 107 TCID50/ml were 10-fold serially diluted in minimum essential medium and subjected to RNA extraction and RT-PCR testing in triplicate for each dilution.
Twenty FMDV strains, two SVDV strains, six VSV strains, two VESV strains, and four CSFV strains (Table 2) were used for evaluating the analytical specificities of the SVV RT-PCRs in the National Centre for Foreign Animal Disease (NCFAD) located at Winnipeg, Canada. The FMDV and SVDV isolates were obtained by NCFAD from the World Reference Laboratory for FMD, Pirbright Institute, UK which usually receives FMDV submissions from affected countries. At the NCFAD, FMDV and SVDV isolates were propagated in baby hamster kidney (BHK-21, ATCC CCL-10) and porcine kidney (IBRS2, ATCC CRL-1835) cell lines, respectively. The VSV isolates were obtained from Plum Island Animal Disease Center, Greenport, NY, USA and amplified in African green monkey (Vero, ATCC CCL-81) cells at the NCFAD. The CSFV and VESV isolates were obtained by the NCFAD respectively from the World Reference Laboratory for CSF in Hannover, Germany and Animal Disease Research Institute, Nepean, Canada and each was amplified in porcine kidney (PK-15, CCL-33) cells. Nucleic acid was extracted from the cell culture supernatants for specificity testing.
Additional swine viral pathogens used for evaluating analytical specificities of the SVV RT-PCRs included PEDV, PDCoV, transmissible gastroenteritis virus, porcine rotaviruses (A, B, C), porcine parvovirus 1, porcine circovirus 2, PRRSV-1, PRRSV-2, swine influenza A virus (H1N1 and H3N2), porcine hemagglutinating encephalomyelitis virus, porcine respiratory coronavirus, pseudorabies virus, porcine teschovirus, and porcine sapelovirus (Table 1). All of these viruses were available at the ISU VDL and were grown in the appropriate cell lines obtained from ATCC. Phosphate buffered saline (PBS) was included as a negative control. Nucleic acid was extracted from these viruses and PBS for specificity testing.
Clinical samples
A total of 125 swine clinical samples (12 vesicular swabs, 30 tonsil swabs, 25 oral fluids, 28 sera, and 30 fecal swabs) collected from various states within the U.S. since 2015 were used to evaluate the diagnostic performances of SVV RT-PCRs. All of these samples were submitted by veterinarians to the Iowa State University Veterinary Diagnostic Laboratory for routine testing.
Nucleic acid extraction
Nucleic acids were extracted from various virus isolates, vesicular swabs and sera using a volume of 50 μL of samples as well as from tonsil swabs, oral fluids and fecal swabs using a volume of 100 μL of samples. Nucleic acid extraction was conducted using a MagMAX™ Pathogen RNA/DNA Kit (Thermo Fisher Scientific, Waltham, Massachusetts, USA) on a Kingfisher-Flex instrument (Thermo Fisher Scientific) following the instructions of the manufacturer. Nucleic acids were eluted into 90 μL of Elution buffer.
Nucleic acids were also extracted from clinical samples (vesicular swabs, tonsil swabs, oral fluids, sera and fecal swabs) using taco™ Preloaded DNA/RNA Extraction Kit on a taco™ mini. Briefly, 200 μl of the samples were added into the first wells of an extraction plate which was subsequently placed into the device following the manufacturer’s user manual. Nucleic acids were eluted into 200 μl of Elution buffer.
In vitro transcribed (IVT) RNA
To prepare the SVV RNA standards, plasmids containing a fragment of 5′ UTR or 3D region of SVV USA/SD41901/2015 (GenBank accession no. KT757281) were synthesized (IDT, Coralville, Iowa, USA). The plasmids were linearized, purified and subjected to run-off in vitro transcription into RNA using a MEGAscript T7 Transcription Kit (Thermo Fisher Scientific). RNA transcripts were produced, treated with Turbo DNase, and purified using the MEGAclear™ Transcription Clean-Up Kit (Thermo Fisher Scientific) following the manufacturer’s instructions. Copy numbers of RNA transcripts were calculated based on concentrations determined by a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). Serial dilutions of RNA were prepared in nucleic acid dilution solution. Aliquots were frozen at − 80 °C for single use of each aliquot.
SVV real-time RT-PCR and SVV RT-iiPCR
SVV rRT-PCR was set up in a 20 μL total reaction using TaqMan® Fast 1-Step Master Mix (Thermo Fisher Scientific): 5 μL of 4× Master Mix, 0.4 μL of forward primer at 20 μM, 0.4 μL of reverse primer at 20 μM, 0.24 μL of probe at 10 μM, 1 μL XENO Internal Positive Control Reagent (Thermo Fisher Scientific), 7.96 μL nuclease-free water, and 5 μL nucleic acid extract. Amplification reactions were performed on an ABI 7500 Fast instrument (Thermo Fisher Scientific) with the following conditions: 1 cycle of 50 °C for 5 min, 1 cycle of 95 °C for 20 s, and 40 cycles of 95 °C for 3 s and 60 °C for 30 s. Any cycle threshold (CT) value < 40 was reported as positive.
The POCKIT™ SVV Reagent Set (GeneReach USA) was a lyophilized TaqMan probe-based RT-PCR reaction. The Premix was reconstituted with 50 μL Premix Buffer B before the addition of 5 μL nucleic acid, and 50 μL of the mixture was transferred to an R-tube™ (GeneReach USA). The tube was spun briefly in a cubee™ microcentrifuge and placed into a POCKIT™ Nucleic Acid Analyzer for RT-PCR reaction. The default program in the POCKIT™ device converted automatically the signal-to-noise (S/N) ratio to positive “+” or negative “-” [36] and display them on the screen after the reaction is completed. Based on the default thresholds, S/N ratios of < 1.2 and > 1.3 were assigned as “+” and “−”, respectively. A “?” result was assigned to those with an S/N ratio between 1.2 and 1.3, indicating that the signals were ambiguous and the sample should be retested again.
Statistical analyses
Limit of detection 95% (LoD95%) was determined by probit analysis with a 95% confidence interval (95% CI) by using the SPSS v14 (SPSS, Chicago, IL, USA). Kappa analyses was used to assess interrater agreement.
Availability of data and materials
The data set(s) supporting the results of this article are included within the article.
Abbreviations
- CSFV:
-
Classical swine fever virus
- CT :
-
Cycle threshold
- FMDV:
-
Foot-and-mouth disease virus
- IVT:
-
In vitro transcribed
- LoD:
-
Limit of detection
- PDCoV:
-
Porcine deltacoronavirus
- PEDV:
-
Porcine epidemic diarrhea virus
- PRRSV:
-
Porcine reproductive and respiratory syndrome virus
- rRT-PCR:
-
Real-time RT-PCR
- RT-iiPCR:
-
Reverse transcription insulated isothermal PCR
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- SVDV:
-
Swine vesicular disease virus
- SVV:
-
Seneca Valley virus
- TGEV:
-
Transmissible gastroenteritis virus
- UTR:
-
Untranslated region
- VESV:
-
Vesicular exanthema of swine virus
- VSV:
-
Vesicular stomatitis virus
References
Adams MJ, Lefkowitz EJ, King AM, Bamford DH, Breitbart M, Davison AJ, Ghabrial SA, Gorbalenya AE, Knowles NJ, Krell P, et al. Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses (2015). Arch Virol. 2015;160(7):1837–50.
Hales LM, Knowles NJ, Reddy PS, Xu L, Hay C, Hallenbeck PL. Complete genome sequence analysis of Seneca Valley virus-001, a novel oncolytic picornavirus. J Gen Virol. 2008;89(Pt 5:1265–75.
Knowles NJ, Hales LM, Jones BH, Landgraf JG, House JA, Skele KL, Burroughs KD, Hallenbeck PL. Epidemiology of Seneca Valley virus: identification and characterization of isolates from pigs in the United States. Inari. Abstract G2: The Northern Lights EUROPIC 2006 - 14th Meeting of the European Study Group on the Molecular Biology of Picornaviruses; 2006.
Pasma T, Davidson S, Shaw SL. Idiopathic vesicular disease in swine in Manitoba. Can Vet J. 2008;49(1):84–5.
Singh K, Corner S, Clark SG, Scherba G, Fredrickson R. Seneca Valley virus and vesicular lesioins in a pig with idiopathic vesicular disease. J Vet Sci Technol. 2012;3:123.
Leme RA, Zotti E, Alcantara BK, Oliveira MV, Freitas LA, Alfieri AF, Alfieri AA. Senecavirus a: an emerging vesicular infection in Brazilian pig herds. Transbound Emerg Dis. 2015;62(6):603–11.
Vannucci FA, Linhares DC, Barcellos DE, Lam HC, Collins J, Marthaler D. Identification and complete genome of Seneca Valley virus in vesicular fluid and sera of pigs affected with idiopathic vesicular disease, Brazil. Transbound Emerg Dis. 2015;62(6):589–93.
Leme RA, Oliveira TE, Alcantara BK, Headley SA, Alfieri AF, Yang M, Alfieri AA. Clinical manifestations of Senecavirus a infection in neonatal pigs, Brazil, 2015. Emerg Infect Dis. 2016;22(7):1238–41.
Leme RA, Alfieri AF, Alfieri AA. Update on Senecavirus infection in pigs. Viruses. 2017;9(7):E170.
Zhang J, Pineyro P, Chen Q, Zheng Y, Li G, Rademacher C, Derscheid R, Guo B, Yoon KJ, Madson D, et al. Full-length genome sequences of Senecavirus a from recent idiopathic vesicular disease outbreaks in U.S. swine. Genome Announc. 2015;3(6):e01270–15.
Guo B, Pineyro PE, Rademacher CJ, Zheng Y, Li G, Yuan J, Hoang H, Gauger PC, Madson DM, Schwartz KJ, et al. Novel Senecavirus a in swine with vesicular disease, United States, July 2015. Emerg Infect Dis. 2016;22(7):1325–7.
Canning P, Canon A, Bates JL, Gerardy K, Linhares DC, Pineyro PE, Schwartz KJ, Yoon KJ, Rademacher CJ, Holtkamp D, et al. Neonatal mortality, vesicular lesions and lameness associated with Senecavirus a in a U.S. sow farm. Transbound Emerg Dis. 2016;63(4):373–8.
Gimenez-Lirola LG, Rademacher C, Linhares D, Harmon K, Rotolo M, Sun Y, Baum DH, Zimmerman J, Pineyro P. Serological and molecular detection of Senecavirus a associated with an outbreak of swine idiopathic vesicular disease and neonatal mortality. J Clin Microbiol. 2016;54(8):2082–9.
Hause BM, Myers O, Duff J, Hesse RA. Senecavirus a in pigs, United States, 2015. Emerg Infect Dis. 2016;22(7):1323–5.
Joshi LR, Mohr KA, Clement T, Hain KS, Myers B, Yaros J, Nelson EA, Christopher-Hennings J, Gava D, Schaefer R, et al. Detection of the emerging picornavirus Senecavirus a in pigs, mice, and houseflies. J Clin Microbiol. 2016;54(6):1536–45.
Wang L, Prarat M, Hayes J, Zhang Y. Detection and genomic characterization of Senecavirus a, Ohio, USA, 2015. Emerg Infect Dis. 2016;22(7):1321–3.
Wu Q, Zhao X, Bai Y, Sun B, **e Q, Ma J. The first identification and complete genome of Senecavirus a affecting pig with idiopathic vesicular disease in China. Transbound Emerg Dis. 2017;64(5):1633–40.
Wang H, Li C, Zhao B, Yuan T, Yang D, Zhou G, Yu L. Complete genome sequence and phylogenetic analysis of Senecavirus a isolated in Northeast China in 2016. Arch Virol. 2017;162(10):3173–6.
Zhang X, **ao J, Ba L, Wang F, Gao D, Zhang J, Pan C, Qi P. Identification and genomic characterization of the emerging Senecavirus a in Southeast China, 2017. Transbound Emerg Dis. 2018;65(2):297–302.
Zhao X, Wu Q, Bai Y, Chen G, Zhou L, Wu Z, Li Y, Zhou W, Yang H, Ma J. Phylogenetic and genome analysis of seven senecavirus a isolates in China. Transbound Emerg Dis. 2017;64(6):2075–82.
Zhu Z, Yang F, Chen P, Liu H, Cao W, Zhang K, Liu X, Zheng H. Emergence of novel Seneca Valley virus strains in China, 2017. Transbound Emerg Dis. 2017;64(4):1024–9.
Xu W, Hole K, Goolia M, Pickering B, Salo T, Lung O, Nfon C. Genome wide analysis of the evolution of Senecavirus a from swine clinical material and assembly yard environmental samples. PLoS One. 2017;12(5):e0176964.
Saeng-Chuto K, Rodtian P, Temeeyasen G, Wegner M, Nilubol D. The first detection of Senecavirus a in pigs in Thailand, 2016. Transbound Emerg Dis. 2018;65(1):285–8.
Sun D, Vannucci F, Knutson TP, Corzo C, Marthaler DG. Emergence and whole-genome sequence of Senecavirus a in Colombia. Transbound Emerg Dis. 2017;64(5):1346–9.
Montiel N, Buckley A, Guo B, Kulshreshtha V, VanGeelen A, Hoang H, Rademacher C, Yoon KJ, Lager K. Vesicular disease in 9-week-old pigs experimentally infected with Senecavirus a. Emerg Infect Dis. 2016;22(7):1246–8.
Joshi LR, Fernandes MH, Clement T, Lawson S, Pillatzki A, Resende TP, Vannucci FA, Kutish GF, Nelson EA, Diel DG. Pathogenesis of Senecavirus a infection in finishing pigs. J Gen Virol. 2016;97(12):3267–79.
Fernandes MHV, Maggioli MF, Joshi LR, Clement T, Faccin TC, Rauh R, Bauermann FV, Diel DG. Pathogenicity and cross-reactive immune responses of a historical and a contemporary Senecavirus a strains in pigs. Virology. 2018;522:147–57.
Feronato C, Leme RA, Diniz JA, Agnol AMD, Alfieri AF, Alfieri AA. Development and evaluation of a nested-PCR assay for Senecavirus a diagnosis. Trop Anim Health Prod. 2018;50(2):337–44.
Bracht AJ, O'Hearn ES, Fabian AW, Barrette RW, Sayed A. Real-time reverse transcription PCR assay for detection of Senecavirus a in swine vesicular diagnostic specimens. PLoS One. 2016;11(1):e0146211.
Fowler VL, Ransburgh RH, Poulsen EG, Wadsworth J, King DP, Mioulet V, Knowles NJ, Williamson S, Liu X, Anderson GA, et al. Development of a novel real-time RT-PCR assay to detect Seneca Valley virus-1 associated with emerging cases of vesicular disease in pigs. J Virol Methods. 2017;239:34–7.
Dall Agnol AM, Otonel RAA, Leme RA, Alfieri AA, Alfieri AF. A TaqMan-based qRT-PCR assay for Senecavirus a detection in tissue samples of neonatal piglets. Mol Cell Probes. 2017;33:28–31.
Pinheiro-de-Oliveira TF, Fonseca-Junior AA, Camargos MF, Laguardia-Nascimento M, Giannattasio-Ferraz S, Cottorello ACP, de Oliveira AM, Goes-Neto A, Barbosa-Stancioli EF. Reverse transcriptase droplet digital PCR to identify the emerging vesicular virus Senecavirus a in biological samples. Transbound Emerg Dis. 2019;66(3):1360–69.
Zhang Z, Zhang Y, Lin X, Chen Z, Wu S. Development of a novel reverse transcription droplet digital PCR assay for the sensitive detection of Senecavirus a. Transbound Emerg Dis. 2019;66(1):517–25.
Zeng F, Cong F, Liu X, Lian Y, Wu M, **ao L, Yuan W, Huang R, Ma J, Guo P, et al. Development of a real time loop-mediated isothermal amplification method for detection of Senecavirus a. J Virol Methods. 2018;261:98–103.
Armson B, Walsh C, Morant N, Fowler VL, Knowles NJ, Clark D. The development of two field-ready reverse transcription loop-mediated isothermal amplification assays for the rapid detection of Seneca Valley virus 1. Transbound Emerg Dis. 2019;66(1):497–504.
Tsai YL, Wang HT, Chang HF, Tsai CF, Lin CK, Teng PH, Su C, Jeng CC, Lee PY. Development of TaqMan probe-based insulated isothermal PCR (iiPCR) for sensitive and specific on-site pathogen detection. PLoS One. 2012;7(9):e45278.
Chang HF, Tsai YL, Tsai CF, Lin CK, Lee PY, Teng PH, Su C, Jeng CC. A thermally baffled device for highly stabilized convective PCR. Biotechnol J. 2012;7(5):662–6.
Lung O, Pasick J, Fisher M, Buchanan C, Erickson A, Ambagala A. Insulated isothermal reverse transcriptase PCR (iiRT-PCR) for rapid and sensitive detection of classical swine fever virus. Transbound Emerg Dis. 2016;63(5):e395–402.
Ambagala A, Fisher M, Goolia M, Nfon C, Furukawa-Stoffer T, Ortega Polo R, Lung O. Field-deployable reverse transcription-insulated isothermal PCR (RT-iiPCR) assay for rapid and sensitive detection of foot-and-mouth disease virus. Transbound Emerg Dis. 2017;64(5):1610–23.
Zhang J, Tsai YL, Lee PY, Chen Q, Zhang Y, Chiang CJ, Shen YH, Li FC, Chang HF, Gauger PC, et al. Evaluation of two singleplex reverse transcription-insulated isothermal PCR tests and a duplex real-time RT-PCR test for the detection of porcine epidemic diarrhea virus and porcine deltacoronavirus. J Virol Methods. 2016;234:34–42.
Kuo HC, Lo DY, Chen CL, Lee CH, Shen YH, Tsai YL, Lee PY, Chang HF. Rapid detection of type 2 porcine reproductive and respiratory syndrome virus by a duplex reverse transcription insulated isothermal PCR on a field-deployable system. J Vet Sci Technol. 2017;8:415.
Tsai YL, Lin YC, Chou PH, Teng PH, Lee PY. Detection of white spot syndrome virus by polymerase chain reaction performed under insulated isothermal conditions. J Virol Methods. 2012;181(1):134–7.
Tsai YL, Wang HC, Lo CF, Tang-Nelson K, Lightner D, Ou BR, Hour AL, Tsai CF, Yen CC, Chang HF, et al. Validation of a commercial insulated isothermal PCR-based POCKIT test for rapid and easy detection of white spot syndrome virus infection in Litopenaeus vannamei. PLoS One. 2014;9(3):e90545.
Wilkes RP, Tsai YL, Lee PY, Lee FC, Chang HF, Wang HT. Rapid and sensitive detection of canine distemper virus by one-tube reverse transcription-insulated isothermal polymerase chain reaction. BMC Vet Res. 2014;10:213.
Wilkes RP, Lee PY, Tsai YL, Tsai CF, Chang HH, Chang HF, Wang HT. An insulated isothermal PCR method on a field-deployable device for rapid and sensitive detection of canine parvovirus type 2 at points of need. J Virol Methods. 2015;220:35–8.
Wilkes RP, Kania SA, Tsai YL, Lee PY, Chang HH, Ma LJ, Chang HF, Wang HT. Rapid and sensitive detection of feline immunodeficiency virus using an insulated isothermal PCR-based assay with a point-of-need PCR detection platform. J Vet Diagn Investig. 2015;27(4):510–5.
Wilkes RP, Anis E, Dunbar D, Lee PA, Tsai YL, Lee FC, Chang HG, Wang HT, Graham EM. Rapid and sensitive insulated isothermal PCR for point-of-need feline leukaemia virus detection. J Feline Med Surg. 2018;20(4):362–9.
Kuo HC, Lo DY, Chen CL, Tsai YL, ** JF, Lee CH, Lee PA, Chang HG. Rapid and sensitive detection of mycoplasma synoviae by an insulated isothermal polymerase chain reaction-based assay on a field-deployable device. Poult Sci. 2017;96(1):35–41.
Soltan MA, Tsai YL, Lee PA, Tsai CF, Chang HG, Wang HT, Wilkes RP. Comparison of electron microscopy, ELISA, real time RT-PCR and insulated isothermal RT-PCR for the detection of rotavirus group a (RVA) in feces of different animal species. J Virol Methods. 2016;235:99–104.
Balasuriya UB, Lee PY, Tiwari A, Skillman A, Nam B, Chambers TM, Tsai YL, Ma LJ, Yang PC, Chang HF, et al. Rapid detection of equine influenza virus H3N8 subtype by insulated isothermal RT-PCR (iiRT-PCR) assay using the POCKIT nucleic acid analyzer. J Virol Methods. 2014;207:66–72.
Carossino M, Lee PY, Nam B, Skillman A, Shuck KM, Timoney PJ, Tsai YL, Ma LJ, Chang HF, Wang HT, et al. Development and evaluation of a reverse transcription-insulated isothermal polymerase chain reaction (RT-iiPCR) assay for detection of equine arteritis virus in equine semen and tissue samples using the POCKIT system. J Virol Methods. 2016;234:7–15.
Balasuriya UB, Lee PA, Tsai YL, Tsai CF, Shen YH, Chang HG, Skillman A, Wang HT, Pronost S, Zhang Y. Translation of a laboratory-validated equine herpesvirus-1 specific real-time PCR assay into an insulated isothermal polymerase chain reaction (iiPCR) assay for point-of-need diagnosis using POCKIT nucleic acid analyzer. J Virol Methods. 2017;241:58–63.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Acknowledgements
All of the clinical swine samples used in this study were submitted by veterinarians to the Iowa State University Veterinary Diagnostic Laboratory for routine testing. We appreciate veterinarians for submitting samples to the Iowa State University Veterinary Diagnostic Laboratory.
Funding
This project was partially funded by the Swine Health Information Center (Project #16–271 SHIC). The funding agency has no role in the design of the study, in collection, analysis, and interpretation, and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: JZ, PYL, and HTW. Designed primers and probes: KH, CHL, and YLT. Prepared virus isolates and clinical samples: CN, LF, QC, and PP. Cloned and prepared in vitro transcribed RNA: AS and CHL. Performed SVV rRT-PCR and RT-iiPCR: SB, CN, CFT, and CHL. Conducted data analysis: CHL, CFT and JZ. Prepared and revised the manuscript: JZ, PYL, CN, CFT, CHL, LF, QC, AS, SB, KH, PP, PG, YLT, and HTW. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable. All of the clinical swine samples used in this study were submitted by veterinarians to the Iowa State University Veterinary Diagnostic Laboratory for routine testing.
Consent for publication
Not applicable.
Competing interests
Chuan-Fu Tsai, Chien-Hsien Lee, Yun-Long Tsai, Hwa-Tang Thomas Wang, and Pei-Yu Alison Lee were employees of GeneReach USA at the time this study was performed. However, this does not alter our adherence to all the BMC Veterinary Research’s policies on sharing data and materials. The other authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, J., Nfon, C., Tsai, CF. et al. Development and evaluation of a real-time RT-PCR and a field-deployable RT-insulated isothermal PCR for the detection of Seneca Valley virus. BMC Vet Res 15, 168 (2019). https://doi.org/10.1186/s12917-019-1927-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-019-1927-4