
Abstract
Background
SWI/SNF (BAF) chromatin remodeling complexes regulate lineage-specific enhancer activity by promoting accessibility for diverse DNA-binding factors and chromatin regulators. Additionally, they are known to modulate the function of the epigenome through regulation of histone post-translational modifications and nucleosome composition, although the way SWI/SNF complexes govern the epigenome remains poorly understood. Here, we investigate the function of ARID1A, a subunit of certain mammalian SWI/SNF chromatin remodeling complexes associated with malignancies and benign diseases originating from the uterine endometrium.
Results
Through genome-wide analysis of human endometriotic epithelial cells, we show that more than half of ARID1A binding sites are marked by the variant histone H3.3, including active regulatory elements such as super-enhancers. ARID1A knockdown leads to H3.3 depletion and gain of canonical H3.1/3.2 at ARID1A-bound active regulatory elements, and a concomitant redistribution of H3.3 toward genic elements. ARID1A interactions with the repressive chromatin remodeler CHD4 (NuRD) are associated with H3.3, and ARID1A is required for CHD4 recruitment to H3.3. ZMYND8 interacts with CHD4 to suppress a subset of ARID1A, CHD4, and ZMYND8 co-bound, H3.3+ H4K16ac+ super-enhancers near genes governing extracellular matrix, motility, adhesion, and epithelial-to-mesenchymal transition. Moreover, these gene expression alterations are observed in human endometriomas.
Conclusions
These studies demonstrate that ARID1A-containing BAF complexes are required for maintenance of the histone variant H3.3 at active regulatory elements, such as super-enhancers, and this function is required for the physiologically relevant activities of alternative chromatin remodelers.
Similar content being viewed by others


Background
The SWI/SNF complex remodels chromatin through ATP-dependent DNA sliding, H2A/H2B dimer eviction, and nucleosome ejection functions [1,2,3]. SWI/SNF remodeling activities open chromatin and promote accessibility for other DNA-binding factors and chromatin regulators [4, 5]. SWI/SNF complex composition is heterogeneous and cell type dependent [6]. SWI/SNF regulates lineage-specific enhancer activity through multiple mechanisms [7]. Protein subunit architecture contributes to SWI/SNF complex specificity through specialized cofactor interactions. The activities of chromatin remodelers and associated machinery are known to modulate the epigenome by regulating histone post-translational modifications and nucleosome composition [5]. Multiple chromatin remodeler complexes are often observed at the same genomic loci and can perform redundant, cooperative, or antagonistic transcriptional regulatory roles [8].
Subunits within the mammalian SWI/SNF (BAF) chromatin remodeler complex are mutated across an estimated 20% of all human cancers [9]. Tissue-specific propensities for mutations in certain SWI/SNF subunits are also evident [10]. ARID1A (BAF250A) is the most frequently mutated SWI/SNF subunit [11]. ARID1A is the largest SWI/SNF subunit and acts as a structural scaffold for other subunits in certain SWI/SNF complexes [12, 13]. ARID1A also exhibits essential DNA-binding activity albeit in a non-sequence-specific manner [14, 15]. Defects in chromatin accessibility and higher-order chromatin structure are thought to underlie ARID1A and SWI/SNF mutant pathogenesis at least partially [16, 17]. Uterine endometrial cancer displays high rates of ARID1A mutation, with roughly 40% of cases showing loss of ARID1A expression [18, 19]. ARID1A mutations and loss of expression are also observed in deeply invasive forms of endometriosis, which is characterized by ectopic spread of the endometrium [20,21,22]. ARID1A mutations are also common in endometriosis-associated ovarian cancers [23, 24].
In the endometrial epithelium, we have previously shown that ARID1A normally promotes epithelial identity by repressing the expression of mesenchymal and invasion genes, through promoter-proximal and distal chromatin interactions that affect transcriptional activity [25,26,27]. In particular, we have found that ARID1A-dependent repression of super-enhancer activity plays critical roles in the maintenance of epithelial identity in the endometrium [25,26,27]. Other reports have demonstrated that ARID1A and SWI/SNF can function as a repressor, often through interactions with repressive machinery [15, 28,29,30]. Although nucleosome structure and histone post-translational modifications are suspected mechanisms, it remains poorly understood how SWI/SNF governs the epigenome.
Here, we reveal a mechanism by which ARID1A maintains histone variant H3.3 in active chromatin. This regulation is required for binding of the SWI/SNF-like CHD4 (NuRD) remodeler complex and linked to the CHD4-interacting multivalent histone reader ZMYND8, notably at a subset of super-enhancers. We finally reveal that this mechanism of ARID1A, H3.3, CHD4, and ZMYND8 co-repression targets physiologically relevant genes involved in epithelial-to-mesenchymal transition (EMT) and cellular invasion, and these genes are aberrantly upregulated in human endometriomas. Altogether, our studies reveal a role for ARID1A-containing SWI/SNF complexes in the maintenance of H3.3, and, at a subset of physiologically relevant target genes, H3.3, ARID1A, CHD4, and ZMYND8 are required for transcriptional repression.
Results
ARID1A regulates H3.3-associated active chromatin
Our previous studies have demonstrated that ARID1A promotes epithelial characteristics in immortalized 12Z human endometriotic epithelial cells at both the transcriptional and phenotypic levels, such that ARID1A loss leads to epithelial-to-mesenchymal transition (EMT) and enhanced migration and invasion [25, 27]. ARID1A loss in 12Z recapitulates many of the molecular and cellular features observed in ARID1A-deficient endometrial epithelia in vivo [25, 27]. Altogether, 12Z cells represent a model system to explore physiological roles for ARID1A in epigenomic regulation.
Histone H3.3 is a variant of canonical H3 with known ties to active chromatin and transcriptional regulation [31, 32]. Like ARID1A, H3.3 has also been observed to mark and regulate active enhancers [33,34,35,36]. We investigated the relationship between ARID1A binding and H3.3 in 12Z cells. To measure genome-wide H3.3 localization, we performed H3.3 chromatin immunoprecipitation followed by sequencing (ChIP-seq) in control 12Z cells (n = 2 IP replicates). Significant H3.3 enrichment was observed at 40,006 genomic regions (Fig. 1A). Intronic, intergenic, and promoter-TSS regions comprised the vast majority of H3.3 enrichment sites (Fig. 1A). H3.3 ChIP-seq peaks were 1830 bp in width on average and ranged from <500 bp to >10 kilobases (Fig. 1B). Intersecting H3.3 ChIP-seq peaks with our previously published ARID1A ChIP-seq data from these cells [25] revealed that over half of each peak set overlapped (Fig. 1C), a 16.9-fold over-representation genome-wide (Fig. 1D).

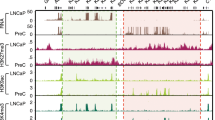
Genome-wide analysis of H3.3-ARID1A chromatin co-regulation. A Genomic annotation of 40,006 genome-wide H3.3 ChIP-seq peaks in 12Z cells (n = 2). B Distribution of H3.3 peak widths. Median H3.3 peak width is 1830 bp. C Genome-wide overlap of ARID1A and H3.3 ChIP-seq peaks. D Genome-wide association between H3.3 and other previously measured chromatin features, per genomic bp, quantified as [observed / expected]. Statistic is hypergeometric enrichment. E Enrichment for H3.3 and ARID1A co-regulation across 18 chromatin states previously modeled via ChromHMM [27]. Left, enrichment of H3.3 peaks; center, enrichment of H3.3+ARID1A binding; right, enrichment of ARID1A binding at sites with vs. without H3.3. Statistic is hypergeometric enrichment. F Left, ARID1A binding levels (ChIP/input fold-enrichment, FE) at H3.3+ vs. H3.3− ARID1A peaks. Right, H3.3 abundance (ChIP/input fold-enrichment) at ARID1A+ vs. ARID1A− H3.3 peaks. Statistic is two-tailed, unpaired Wilcoxon’s test. G Top, enrichment of H3.3 at genes promoter-proximally bound by ARID1A. Bottom, enrichment of ARID1A+H3.3 co-binding at genes DE following ARID1A loss (siARID1A treatment). Statistics are hypergeometric enrichment test and pairwise two-tailed Fisher’s exact test. H Example hg38 browser shots of genes and regulatory elements co-regulated by H3.3 and ARID1A. y-axis is log-likelihood ratio (logLR) of assay signal (compared to input chromatin for ChIP-seq or background genome for ATAC-seq). Small bars under tracks indicate significant peak detection by MACS2 (FDR < 0.05). Super-enhancers were detected by ROSE from H3K27ac ChIP-seq. * p < 0.05, ** p < 0.01, *** p < 0.001
We previously constructed a genome-wide chromatin state map accompanying ARID1A loss in 12Z cells via chromHMM [37] by measuring seven chromatin features associated with transcriptional regulation: total RNA, ATAC (accessibility), H3K27ac, H3K18ac, H3K4me1, H3K4me3, and H3K27me3 [27]. Similar to our previous reports of ARID1A regulated chromatin states, genomic H3.3 enrichment was highly associated with all active, euchromatic features, but not heterochromatic H3K27me3 (Fig. 1D). Annotating H3.3 enrichment in each of our characterized chromatin states revealed that H3.3 is associated with similar regulatory chromatin states as ARID1A binding, most notably super-enhancers and active typical enhancers (Fig. 1E, left). In agreement, co-regulation by H3.3 and ARID1A was most prominently observed at these same chromatin states (Fig. 1E, center). Next, we examined ARID1A binding at H3.3-marked vs. H3.3-absent chromatin sub-states and found that ARID1A binding was associated with H3.3 at promoter-proximal and genic super-enhancers and active transcription start sites (TSS) (Fig. 1E, right). Upon further investigation of ARID1A and H3.3 genome-wide binding patterns, we observed that genome-wide ARID1A peaks showed overall stronger ARID1A binding when H3.3 was also localized, and H3.3 was overall more abundant at genome-wide H3.3 peaks also bound by ARID1A (Fig. 1F). These data indicate that ARID1A and H3.3 may co-regulate active chromatin elements like enhancers and gene promoters.
We previously reported that ARID1A chromatin binding near gene promoters is associated with transcriptional regulation, such that ARID1A loss leads to aberrant gene expression [25]. Our H3.3 data further revealed that ARID1A binding at promoter-proximal regulatory elements is highly enriched among genes marked by promoter-proximal H3.3 (Fig. 1G, top), indicating that ARID1A transcriptional regulation may be coupled with H3.3. Moreover, the 2037 genes co-marked by ARID1A and H3.3 in the promoter-proximal region (±3 kb surrounding TSS) were more likely to show differential expression (DE) following ARID1A loss than genes without promoter-proximal H3.3 (Fig. 1G, bottom). In addition, locus-scale investigation clearly showed that ARID1A and H3.3 often co-mark active chromatin regulatory elements, which infrequently also includes gene body coating by H3.3, such as at COL1A1, THBS1, and SERPINE1 (Fig. 1H). These data collectively suggest H3.3 may be linked to transcriptional regulatory activity by ARID1A at the level of chromatin.
ARID1A chromatin interactions maintain H3.3
To understand the relationship between ARID1A and H3.3, we depleted ARID1A from 12Z cells using lentiviral shRNA particles targeting ARID1A (shARID1A) then measured H3.3 by ChIP-seq. Our differential H3.3 ChIP-seq analysis (shARID1A vs. non-targeting shRNA control, n = 2) indicated that nearly 1/3 of tested H3.3 regions showed significant differences in H3.3 abundance (csaw/edgeR, FDR < 0.05) at 72 h following ARID1A knockdown (Fig. 2A, Additional file 1: Fig. S1A). We noted that ARID1A knockdown in 12Z cells did not result in obvious changes in global H3.3 levels by immunoblotting of the histone fraction (Additional file 1: Fig. S1B), suggesting any effects are likely occurring at the level of chromatin. This is further supported by our previously reported 12Z ARID1A knockdown RNA-seq data [25] indicating that the dominantly expressed H3.3-encoding gene isoform, H3F3B, does not change in expression (Additional file 1: Fig. S1C). We then investigated how ARID1A chromatin binding may be directly associated with the observed changes in H3.3 following ARID1A loss. Strikingly, ARID1A-bound differential H3.3 regions almost exclusively lost H3.3 and rarely gained H3.3 (Fig. 2B). Corroborating this result, we also profiled canonical H3.1/3.2 histone levels by ChIP-seq (n = 2) and observed that ARID1A-bound, H3.3-marked chromatin regions gain H3.1/3.2 (Fig. 2C). While 33% of all tested H3.3 regions had detectable ARID1A binding, 81% of the 8418 shARID1A decreasing H3.3 regions were normally bound by ARID1A, as opposed to only 3% of the 11,059 shARID1A increasing H3.3 regions (Fig. 2D). These results indicate that ARID1A interactions with H3.3 chromatin may serve to promote H3.3 incorporation or maintain its stability. When ARID1A is mutated, H3.3-marked regions shift toward canonical H3.1/3.2.

Genome-wide analysis of ARID1A-dependent H3.3. A MA plot of shARID1A vs. control differential H3.3 ChIP-seq (n = 2), across 67,502 tested genomic regions. Regions are colored based on shARID1A differential H3.3 significance. Inset pie chart depicts distribution of significantly increasing and decreasing H3.3 regions (csaw/edgeR FDR < 0.05) compared to stable H3.3 (FDR > 0.05). FDR < 0.05 was used as the significance threshold for all downstream analyses. B shARID1A differential H3.3 regions segregated by detection of ARID1A binding in wild-type cells. Left, MA plot with all genome-wide H3.3 tested regions, colored by ARID1A binding status. Right, box plot quantification of shARID1A log2FC H3.3 abundance, segregated by ARID1A binding status. Statistic is two-tailed, unpaired Wilcoxon’s test. C Analysis of canonical H3 (H3.1/3.2) changes (ChIP-seq, n = 2) at H3.3-marked genomic regions following ARID1A knockdown (shARID1A), segregated by ARID1A binding status as in B. Statistic is two-tailed, unpaired Wilcoxon’s test. D Enrichment of ARID1A binding detection at regions with decreasing H3.3 following ARID1A loss compared to all tested H3.3 regions. Statistics are hypergeometric enrichment test and pairwise two-tailed Fisher’s exact test. E Magnitude of H3.3 change (log2FC) among ARID1A-bound, shARID1A significantly decreasing vs. increasing H3.3 regions. Statistic is two-tailed, unpaired Wilcoxon’s test. F Distribution of H3.3-enriched region widths among shARID1A stable vs. increasing vs. decreasing H3.3 regions. Statistic is two-tailed, unpaired Wilcoxon’s test. G Chromatin state enrichment among shARID1A increasing and decreasing H3.3 regions, calculated per 200 bp genomic interval. Statistic is hypergeometric enrichment. H Top 10 significant (FDR < 0.05) enriched Hallmark pathways (left) and GO Biological Process gene sets (right) among genes with ARID1A-bound, shARID1A decreasing promoter-proximal H3.3. I Representative hg38 locus near CCL2 displaying H3.3 maintained by ARID1A chromatin interactions. *** p < 0.001
We further characterized the changes in H3.3 occurring following ARID1A loss. Globally, we found that typical enhancers (distal regions marked by H3K27ac and ATAC, >3 kb away from a TSS and excluding super-enhancers) were enriched for shARID1A-driven H3.3 alterations as compared to gene promoter-proximal regions and super-enhancers (Additional file 1: Fig. S1D). Intriguingly, gene promoter-proximal regions displayed both decreasing and increasing H3.3, whereas distal typical enhancers and super-enhancers almost exclusively lost H3.3 if significantly affected (Additional file 1: Fig. S1E). Among ARID1A-bound genomic H3.3 regions, shARID1A decreasing H3.3 regions tended to display greater differences in H3.3 abundance than shARID1A increasing H3.3 regions (Fig. 2E), supporting a role for ARID1A in promoting maintenance of H3.3 rather than limiting it. Regions that displayed shARID1A decreasing H3.3 also tended to have overall wider genomic footprints than increasing or stable H3.3 regions (Fig. 2F). In agreement with where ARID1A-H3.3 co-regulation is most frequently observed, chromatin state enrichment analysis indicated that ARID1A loss led to depletion of H3.3 at promoter-proximal super-enhancers and highly active enhancers, while increasing H3.3 was observed over actively transcribed gene bodies (Fig. 2G). From the 412 genes we identified with ARID1A-bound, shARID1A decreasing promoter-proximal H3.3, we found significant enrichment for inflammatory, hypoxia, apoptosis, locomotion, and EMT pathways, such as CCL2 (Fig. 2H,I). These data suggest that ARID1A maintains H3.3 at active regulatory elements such as enhancers and super-enhancers, and, when ARID1A is lost, redistribution of H3.3 occurs toward active genes already marked by H3.3.
H3.3 depletion phenocopies transcriptional effects of ARID1A loss
We next sought to determine the transcriptional consequences of H3.3 loss in endometrial epithelia. We hypothesized that H3F3B could be knocked down to reduce H3.3 levels for acute transcriptome evaluation without impeding cell health (Fig. 3A). Using siRNA targeting H3F3B (siH3F3B), we observed H3.3 depletion by immunoblotting without affecting the cell cycle (Fig. 3B, Additional file 1: Fig. S2A-B). RNA-seq transcriptome analysis (n = 3) 72 h following siRNA transfection showed clear loss of H3F3B expression, but not H3F3A, accompanying 1608 significant DE genes (DESeq2, FDR < 0.001) including those both upregulated (repressed by H3.3) and downregulated (activated by H3.3) (Fig. 3C–E). As expected, we also observed highly significant enrichment for H3.3-dependent transcriptional changes among genes marked by promoter-proximal H3.3 (Additional file 1: Fig. S2C). Similar to our previous observations with acute ARID1A loss [25], depletion of H3.3 led to mostly minor alterations in gene expression, with the majority of DE genes displaying <0.5 log2FC expression change (Fig. 3E). These data indicate H3.3 serves both activating and repressing roles in transcriptional regulation of endometrial epithelial cells.

Transcriptional effects of H3.3 depletion and overlap with ARID1A. A Baseline relative linear expression of H3F3A (H3-3A) and H3F3B (H3-3B) gene isoforms encoding H3.3, as measured by RNA-seq (n = 3). B Western blot for H3.3 and total H3 in control vs. siH3F3B treated cells. C Global transcriptomic effects of 24,192 genes following H3.3 knockdown via siH3F3B treatment (RNA-seq, n = 3). Red dots represent significant DE genes (DESeq2, FDR < 0.001). D Relative linear expression of H3F3A and H3F3B by RNA-seq in control and siH3F3B cells (n = 3). E Volcano plot depicting siH3F3B vs. control differential gene expression (DGE). Top significant genes are labeled. F Significant overlap in DE genes following H3.3 knockdown (siH3F3B) vs. ARID1A knockdown (siARID1A). Statistic is hypergeometric enrichment. G Directional segregation of siH3F3B/siARID1A overlap** DE genes. A positive association is observed by chi-squared test, i.e., genes are more likely to be upregulated or downregulated in both conditions as opposed to antagonistic regulation. H Scatter plot of siH3F3B vs. siARID1A expression log2FC (with shrinkage correction) for all 19,900 transcriptome-wide commonly detected genes. Statistics are Pearson (r) and Spearman (rs) correlation coefficients. Colored dots indicate significant DE genes (FDR < 0.001) in both treatment conditions. I Association between H3.3 transcriptional repression (siH3F3B upregulation) and transcriptional co-regulation by ARID1A (siARID1A DE). Statistic is two-tailed Fisher’s exact test. J Scatter plot of 196 shared DE genes upregulated following knockdown of either H3.3 or ARID1A. These genes are mutually repressed by H3.3 and ARID1A. K Top significant (FDR < 0.05) enriched gene sets among the 196 ARID1A-H3.3 mutually repressed genes among various gene set databases
Comparing the gene expression changes following H3.3 loss with those following ARID1A loss, we observed significant overlap, with 682 shared dysregulated genes (Fig. 3F). These 682 genes were then grouped by direction of change (upregulated vs. downregulated) to identify genes with the same or different expression patterns following ARID1A vs. H3.3 loss. A significant association was observed between the effects of H3.3 and ARID1A loss indicating shared transcriptional consequences (Fig. 3G). Gene expression changes also positively correlated transcriptome-wide (Fig. 3H). Intriguingly, the 682 genes also affected by ARID1A loss were more likely to be transcriptionally repressed by H3.3 (Fig. 3I). In total, 196 genes were identified as mutually repressed by both ARID1A and H3.3, including PLAU, ADAMTS15, C1S, CD82, CCL2, and CLSTN2 (Fig. 3J). In agreement with differential H3.3 patterns, these 196 co-repressed genes were enriched for similar gene sets as observed among the ARID1A-bound, shARID1A decreasing promoter-proximal H3.3 gene set, including EMT, TNFα signaling, estrogen response, apoptosis, adhesion, migration, extracellular matrix, and collagens (Fig. 3K). Altogether, these data suggest that ARID1A and H3.3 co-regulate similar target genes in endometrial epithelial cells. At the chromatin level, depletion or destabilization of H3.3 as a result of ARID1A loss may lead to the upregulation of a physiologically relevant set of EMT and invasion genes.
ARID1A co-regulates H3.3 with CHD4 and ZMYND8
While our data implicate H3.3 in the ARID1A mutant endometrium, few reports have linked SWI/SNF activity to H3.3 containing nucleosomes [38, 39]. To gain insight into factors associated with H3.3 regulation by ARID1A, we used the ReMap2020 database of 165 million peak regions extracted from genome-wide binding assays [40]. For all 1135 transcriptional regulators included in this database, we calculated genome-wide associations for each set of factor peaks with H3.3-marked (H3.3+) vs. H3.3-absent (H3.3−) ARID1A binding. This analysis revealed that two zinc finger MYND-type proteins, ZMYND11 (BS69) and ZMYND8 (PRKCBP1, RACK7), were among the top co-regulators associated with H3.3+ ARID1A chromatin binding (Fig. 4A, OR = 2.93 and 2.43 for ZMYND11 and ZMYND8, respectively). These data suggest that H3.3 regulation by ARID1A may be mediated by these co-regulators. ZMYND11 and ZMYND8 are multivalent chromatin readers that are suggested to function as interfaces between histones and other chromatin regulator complexes like remodelers, writers, and erasers [41, 42]. Both proteins interact with H3/H4 acetylated tails through bromodomains and may show specificity toward or against H3.3-containing nucleosomes [42,1: Fig. S9J-L), including 603 genes affected by each of the four knockdowns (FDR < 0.05) (Fig. 7F, Additional file 1: Fig. S9L). These included 60 genes mutually repressed by ARID1A, CHD4, ZMYND8, and H3.3 (Fig. 7G). These mechanistic co-repressed genes were enriched for EMT, adhesion, development, locomotion, collagens, and extracellular matrix gene sets (Fig. 7H). Further, 68% of these genes were marked by gene body H4K16ac, an enrichment compared to less than half of all expressed genes (Additional file 1: Fig. S10). Two physiologically relevant target genes revealed through integrative epigenomic analysis are PLAU and TRIO, both of which are located within broad H4K16ac+ domains and near active H3.3+ super-enhancers co-bound by ARID1A, CHD4, and ZMYND8 (Fig. 7I). ARID1A loss leads to decreased promoter-proximal H3.3 abundance and transcriptional hyperactivation of PLAU and TRIO (Fig. 7I). We also observed that co-knockdown of ARID1A and CHD4 led to increased induction of PLAU compared to either knockdown separately (Additional file 1: Fig. S11).
ARID1A-H3.3 repressed chromatin targets are aberrantly activated in human endometriomas
Our studies in the 12Z human endometrial epithelial cell line have revealed a mechanism of cooperative regulation by ARID1A, CHD4, and ZMYND8 at H3.3-marked chromatin. To support the relevance of these chromatin regulatory networks on pathologically related gene expression, we utilized a transcriptome expression data set comparing human endometriomas to control endometrial tissue samples [53]. Endometriomas are a result of ectopic spread of endometrial tissue onto the ovary, forming cysts associated with ovarian cancer development [20, 54], and numerous reports have observed high rates of ARID1A mutation or loss of expression in endometriomas [21, 22, 55]. Three ARID1A-H3.3 related gene sets were investigated for relevance in human endometrioma gene expression alterations: (1) ARID1A-bound, shARID1A decreasing promoter-proximal H3.3 genes (n = 412), (2) ARID1A-H3.3 co-repressed genes (i.e., siARID1A/siH3F3B upregulated, FDR < 0.001, n = 196), and (3) ARID1A-H3.3-CHD4-ZMYND8 co-repressed genes (i.e., upregulated with any knockdown, FDR < 0.05, n = 60). We observed significant enrichment for all three of these gene sets among human endometrioma DGE (Fig. 8A, left). Moreover, the overlap** DE genes were more likely to be upregulated in endometriomas than expected by chance, indicating relief of repression is also observed in pathology (Fig. 8A, right). Similarly, examining the endometrioma vs. control endometrium expression log2FC values indicated that each gene set tended to be overall upregulated in the pathological, pre-cancerous state (Fig. 8B). Mechanistic genes aberrantly activated in endometriomas that could be attributed to disruption of ARID1A-H3.3 chromatin repression mechanisms include C1S, SCARB1, GYPC, WWC3, COL6A2, and MAP4K4 (Fig. 8C). Collectively, our data indicate that ARID1A-SWI/SNF maintains the histone variant H3.3 in active regulatory elements, and a subset of physiologically relevant genes are co-regulated by CHD4 and ZMYND8, such that loss of any of these factors leads to alleviation of transcriptional repression and consequential aberrant gene activation in various endometrial disease contexts where ARID1A mutations are thought to drive pathogenesis (Fig. 9).

Mechanistic gene expression alterations in human endometriomas. A Left, enrichment for ARID1A-H3.3 co-repressive chromatin mechanistic gene sets among human endometrioma (ovarian endometriosis) vs. control endometrium DE genes reported by Hawkins et al. [53], compared to all unique measured genes. Right, proportion of overlap** DE genes that are upregulated vs. downregulated in endometriomas, compared to all unique measured genes. Statistic is hypergeometric enrichment. B Box plots displaying endometrioma expression log2FC values for probes annotated to genes within mechanistic gene sets, compared to all measured probes. Statistic is two-tailed, unpaired Wilcoxon’s test. C Relative expression box-dot plots of 6 genes upregulated in endometriomas vs. control endometrium that are co-repressed by ARID1A, H3.3, CHD4, and ZMYND8. Statistic is limma FDR-adjusted p. * p < 0.05, ** p < 0.01, *** p < 0.001

Proposed model of H3.3 chromatin regulation by ARID1A-SWI/SNF and co-regulators. ARID1A and SWI/SNF chromatin remodeling activities are required for H3.3 incorporation or maintenance at certain active regulatory elements across the genome, such as super-enhancers. When ARID1A is mutated or lost, H3.3 maintenance is disrupted, and nucleosome composition shifts toward canonical H3.1/3.2 at ARID1A-bound sites. Consequential to local H3.3 depletion, H3.3 reader factor occupancy is reduced—such as the CHD4-containing NuRD complex—leading to impaired chromatin regulation and aberrant target gene expression. At H3.3+ H4K16ac+ super-enhancer-like elements located promoter-proximally upstream of genes, H3.3 maintenance by ARID1A-SWI/SNF is associated with repression of transcriptional hyperactivation and the NuRD cofactor ZMYND8
Discussion
We have provided evidence that ARID1A functions to maintain the variant histone H3.3 in active regulatory elements. ARID1A loss leads to H3.3 depletion at active enhancers and super-enhancers, due to disrupted ARID1A chromatin interactions, leading to gain of canonical H3.1/3.2 and redistribution of H3.3 toward active genic and transcribed elements. We further showed that this mechanism is largely independent of H3.3-interacting remodeler CHD4-NuRD. Instead, our data suggest that ARID1A-dependent maintenance of H3.3 is required for CHD4-NuRD binding at a subset of enhancers. Therefore, the BAF complex helps to facilitate H3.3 incorporation, and this activity is required for the recruitment of alternative chromatin remodelers and chromatin regulators with unique regulatory activity.
SWI/SNF is thought to eject nucleosomes and open chromatin [2, 4] rather than assemble nucleosomes. SWI/SNF disruption of nucleosomes may be required for H3.3 incorporation and thus coupled to nucleosome assembly. Therefore, we hypothesize that H3.3 regulation by ARID1A-SWI/SNF occurs by ejecting nucleosomes in favor of H3.3 incorporation by other assembly or chaperone factors, such as HIRA, at active regulatory elements [31]. Unlike the DAXX/ATRX complex, which governs H3.3 incorporation at pericentromeric heterochromatin and telomeres and has intrinsic ATP-dependent remodeling activity, HIRA may rely on other chromatin remodeling complexes for its chaperone activity [31]. In the absence of ARID1A-SWI/SNF, H3.3 nucleosome assembly by HIRA may be impeded by the lack of H3.1/3.2 nucleosome remodeling by the BAF complex. The related CHD1 remodeler is known to be required for H3.3 deposition into chromatin in vivo [56], further suggesting a necessary role for SWI/SNF remodeler activity in H3.3 nucleosome assembly. In addition, both FACT and Polybromo-associated Brm (PBAP) complex are thought to facilitate H3.3 incorporation at boundary elements in Drosophila [57]. P400 is another SWI/SNF-like remodeler recently shown to exchange H3.3 nucleosomes that could also possibly collaborate with SWI/SNF [58].
Given previous associations between H3.3 epigenetic memory and cell fate plasticity [32], it is intriguing to consider a role for BAF complex regulation of H3.3 as being a critical determinant of endometrial epithelial cell identity and homeostasis across the menstrual cycle when proliferation and differentiation occur. Further, it remains possible that alternative SWI/SNF complex configurations also participate in H3.3 maintenance, and these complexes could be responsible for H3.3 incorporation at sites unaffected by ARID1A loss.
ARID1A maintenance of H3.3 is associated with genomic interactions with CHD4, a catalytic subunit in the SWI/SNF-like NuRD remodeler complex. As CHD4 knockdown does not lead to the widespread H3.3 depletion observed with ARID1A knockdown, and ARID1A is required for CHD4 recruitment to active regulatory elements, loss of CHD4 co-regulation of H3.3 chromatin is likely the consequence of ARID1A loss. We also observed sub-stoichiometric physical interactions between ARID1A and CHD4, but the significance of direct ARID1A-CHD4 interactions is unclear. CHD4 interactions with histone reader ZMYND8 appear to be associated with further chromatin target regulation specificity, where ZMYND8 may be recruited to H4(K16)ac-marked chromatin through its bromodomain. However, further experimentation, such as ZMYND8 depletion or bromodomain mutation, would be required to confirm the suspected function of the ZMYND8 module in complex recruitment. ZMYND8 co-regulation appears to be associated with chromatin repression, notably at promoter-proximal super-enhancers located upstream of genes, such that disruption of this chromatin mechanism causes relief of repression and subsequent transcriptional hyperactivation. Plasminogen activator urokinase (PLAU) was identified as a key target gene repressed by this mechanism in our 12Z endometriotic epithelial cell model. PLAU was also recently observed as transcriptionally activated during human menstruation [59], suggesting similar repressive chromatin mechanisms may govern PLAU regulation in the healthy endometrium. PLAU is upregulated in ovarian endometrioid carcinomas from women with concurrent endometriosis [60], suggesting PLAU upregulation may promote malignant transformation in endometriosis. ARID1A mutations are frequently observed in endometriosis-associated ovarian cancers [23, 24]. C1S, a component of the complement C1 complex, is another gene that is transcriptionally repressed by ARID1A, CHD4, ZMYND8, and H3.3 that is aberrantly upregulated in human endometriomas. It has been reported that the complement system is activated in women with endometriosis [61], suggesting that ARID1A mutation and associated disruption of chromatin repression may be a possible disease mechanism. In addition to ARID1A, it should be noted that CHD4 mutations leading to nucleosome remodeling defects are also frequent in endometrial cancer [62,63,64] and may lead to de-repression of similar target genes.
H3.3 is considered an active chromatin mark associated with transcriptional activation. However, our data and others have demonstrated that H3.3 can play roles in transcriptional repression, as well as transcriptional poising and higher-order chromatin regulation, although the mechanisms governing these specific activities remain unclear [31]. A simple hypothesized mechanism explaining how H3.3 can function repressively is through associations with CHD4 and the NuRD complex, as we have studied here. Historically, NuRD has been studied as a repressor due to its subunit composition that includes the histone deacetylases HDAC1/2, although activating roles of NuRD are also known [65,66,67]. An early study of H3.3 chromatin dynamics indicated that NuRD components were associated with active regions marked by high H3.3 turnover [68]. More recently, NuRD has been shown to directly interact with H3.3 nucleosomes [48]. The finding that CHD4 recruitment is dependent upon H3.3 maintenance by ARID1A at a subset of enhancers further supports the notion that CHD4 co-repressive activity at these sites is likely a result of H3.3 regulation by the BAF complex. When ARID1A is mutated or lost, an H3.3 to H3.1/3.2 switch may impair CHD4 binding through its normal H3.3 reader function, leading to loss of NuRD HDAC co-repressive activity. Intriguingly, CHD4/NuRD was recently shown to control super-enhancer accessibility and maintain lower acetylation levels through its HDAC activity [69], similar to our findings with ARID1A by antagonizing P300 [27]. In support of our data, the authors observed physical interactions between CHD4 and SWI/SNF. Recently, NuRD and SWI/SNF recruitment to active TSS and enhancers was impaired in H3.3K4A mutant mouse ESCs [39], suggesting that NuRD and SWI/SNF recruitment is dependent on the K4 residues on H3.3.
In silico analyses from the ReMap 2020 transcriptional regulator peak database [40] predicted that ZMYND8 is highly associated with H3.3 chromatin regulation by ARID1A. Here, we detected high stringency physical interactions between CHD4 and ZMYND8 as a possible explanation of this co-regulatory activity, as we have demonstrated that ARID1A maintenance of H3.3 is required for CHD4 binding at enhancers. Others have also reported that ZMYND8 interacts with NuRD in numerous contexts [41, 44, 46, 47]. Intriguingly, one recent study reported that ZMYND8 directly recognizes mutant H3.3G34R [70]. Our data indicate that ZMYND8 links repressive H3.3 to H4 acetylation. In support, the ZMYND8 bromodomain directly interacts with acetylated H4 tails [44], and TIP60-mediated H4 acetylation can functionally recruit ZMYND8 through this mechanism to repress transcription with CHD4 in response to DNA damage [46]. Our data also indicate that ARID1A directly suppresses chromatin accessibility at sites marked by H4 acetylation, suggesting that SWI/SNF chromatin remodeler activity may be involved in ZMYND8-NuRD-mediated chromatin repression. ZMYND8-NuRD repression in response to DNA damage was previously shown to rely on KDM5A demethylase activity [71], further suggesting other factors may orchestrate repression vs. activation logic. ZMYND8 has been reported to be a super-enhancer factor that suppresses hyperactivation [49]. Corroborating our results, ZMYND8 was previously shown to associate with NuRD at super-enhancers [47]. We found that super-enhancers that become hyperacetylated following ARID1A loss are normally associated with the highest levels of H3.3 and ZMYND8 binding. In our proposed model, ZMYND8 bromodomain interactions with H4 acetylated tails might facilitate recruitment and transcriptional repression at active chromatin in association with NuRD, such as at H3.3+ super-enhancers. Further work will seek to elucidate how ZMYND8 functions toward transcriptional repression.
In addition to promoter-proximal and distal enhancer chromatin regulation, SWI/SNF, NuRD, and ZMYND8 have been shown to mediate transcriptional pausing and elongation by Pol II and associated machinery [72,73,74,75], as well as DNA repair [46, 76, 77]. Super-enhancers mark critical cell identity genes [78], and recent evidence suggests chromatin mechanisms coupling transcription and DNA repair occur at super-enhancers to control transcriptional hyperactivation [79]. Super-enhancer chromatin co-regulation by ARID1A, CHD4, and ZMYND8 may fine-tune transcriptional activation states and thus reflect a mechanism at the intersection of transcriptional regulation and other chromatin-regulated processes.
Conclusions
In summary, ARID1A-SWI/SNF activities facilitate maintenance of the histone variant H3.3 in active chromatin, such that ARID1A loss leads to local H3.3 depletion, gain of canonical H3.1/3.2, and H3.3 redistribution toward genic elements with transcriptional consequences. At physiologically relevant genomic regions like super-enhancers, ARID1A collaborates with the repressive CHD4-NuRD remodeling complex and reader protein, ZMYND8, to suppress hyperactivation associated with ARID1A-dependent maintenance of H3.3. ARID1A-CHD-ZMYND8-mediated repression affects genes that are aberrantly activated in human endometriomas. These studies have revealed that SWI/SNF regulation of variant histone exchange influences the activities of other chromatin remodelers and regulators by altering nucleosome substrates, and this mechanism plays substantial roles in women’s health and disease.
Methods
Cell culture, siRNA transfections, and lentiviral shRNA particle usage
Adherent, human 12Z endometriotic epithelial cells were cultured in DMEM/F12 media in the presence of 10% serum (FBS), 1% L-glutamine, and 1% penicillin/streptomycin. Cells were seeded in antibiotic-free media the day before siRNA transfection. Then, 50 nM siRNA (Dharmacon, ON-TARGETplus) were transfected into cells using the Lipofectamine RNAiMAX (Thermo Fisher Scientific) reagent, according to the manufacturer protocol, in OptiMEM (Gibco). Growth media was replaced 24 h following transfection, without antibiotics. Forty-eight hours after transfection, low serum (0.5% FBS) growth media was added with antibiotics. Cells were harvested 72 h following siRNA transfection. Lentiviral shRNA particles were prepared with Lenti-X 293T cells (Takara) and MISSION pLKO.1 plasmids (Sigma-Aldrich) as previously described [27]. Lentiviral shRNA particles were titered using the qPCR Lentiviral Titration Kit (ABM). shRNA particles were transduced into 12Z cells at a 100-fold multiplicity of infection, and media was replaced 24 h later. Cells were harvested 72 h following shRNA transduction.
Cell cycle analysis
The Click-iT Plus EdU Flow cytometry Assay Kit (Invitrogen) was used for cell cycle assays. 12Z cells were treated with 10 mM of EdU for 2 h in culture media. Cells were harvested by trypsinization and washed in 1% BSA in PBS. Cells were resuspended in 100 μL of ice-cold PBS, and 900 μL of ice-cold 70% ethanol was added dropwise while vortexing. Cells were incubated on ice for 2 h. Cells were washed with 1% BSA in PBS and then treated with the Click-iT Plus reaction cocktail including Alexa Fluor 488 picolyl azide according to the manufacturer’s instructions for 30 min. Cells were washed with 1× Click-iT permeabilization buffer and wash reagent, and then treated with 5 mM of Vybrant DyeCycle Ruby Stain (Thermo Fisher) diluted in 1% BSA in PBS for 30 min at 37 °C. Flow cytometry was performed using a BD Accuri C6 flow cytometer (BD Biosciences) and analyzed using FlowJo v10 software (BD Biosciences).
Histone extraction
12Z cells were washed with PBS and scraped in PBS containing 5 mM sodium butyrate. Cells were centrifuged and resuspended in TEB buffer (PBS supplemented with 0.5% Triton X-100, 5 mM sodium butyrate, 2 mM phenylmethylsulfonyl fluoride, 1× protease inhibitor cocktail) and incubated on a 3D spindle nutator at 4 °C for 10 min. Cells were centrifuged at 3000 RPM for 10 min at 4 °C. TEB wash step was repeated once. Following second wash, pellet was resuspended in 0.2 N HCl, and incubated on 3D spindle nutator at 4 °C overnight. The following day, samples were neutralized with 1:10 volume 1M Tris-HCl pH 8.3. Sample was centrifuged at 3000 RPM for 10 min at 4 °C, and supernatant containing histone proteins was collected.
Co-immunoprecipitation (co-IP)
Nuclear extracts were prepared as previously described [15], dialyzed overnight into 0% glycerol (25 mM HEPES, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl, 1 mM DTT) using a Slide-A-Lyzer G2 Dialysis Cassette (10 kDa cutoff, Thermo Fisher Scientific), and quantified with the BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific). Primary antibodies (anti-ARID1A, D2A8U, Cell Signaling; anti-CHD4, D8B12, Cell Signaling) were conjugated to Protein A Dynabeads (Invitrogen) overnight at 4 °C in 1× PBS + 0.5% BSA. Normal rabbit IgG (Cell Signaling) IPs were performed in parallel at equivalent masses, as negative controls. Five hundred micrograms nuclear lyase was diluted into IP buffer (20 mM HEPES, 150 mM KCl, 10% glycerol, 0.2 mM EDTA, 0.1% Tween-20, 0.5 mM DTT) to a final volume of 1 mL and clarified by centrifugation. After overnight IP at 4 °C, bead slurries were washed with a series of IP buffers at different KCl concentrations: 2× washes at 150 mM, 3× washes at 300 mM, 2× washes at 100 mM, 1× wash at 60 mM. Immunoprecipitants were eluted in 2× Laemmli buffer + 100 mM DTT at 70 °C for 10 min with agitation.
Glycerol gradient sedimentation
Nuclear extracts were prepared, dialyzed, and quantified as described in the co-IP methods section. Density sedimentation by glycerol gradient was performed and probed similar to published reports [13]. Briefly, 4.5 mL 10–30% linear glycerol gradients were prepared using an ÄKTA start (Cytiva) from density sedimentation buffer (25 mM HEPES, 0.1 mM EDTA, 12.5 mM MgCl2, 100 mM KCl, 1 mM DTT) additionally containing 30 and 10% glycerol for initial and target concentrations, respectively. Two hundred micrograms nuclear lyase was overlaid on the glycerol gradient followed by ultracentrifugation at 40,000 rpm in an AH-650 swinging bucket rotor (Thermo Fisher Scientific) for 16 h at 4 °C. Two hundred twenty-five microliters gradient fractions were collected and concentrated using StrataClean resin (Agilent). Concentrated fractions were eluted in 1.5× Laemmli buffer + 37.5 mM DTT and run on SDS-PAGE for immunoblotting.
Immunoblotting
Whole-cell protein lysates were prepared as previously described [27]. Proteins were quantified with the BCA Protein Assay Kit (Pierce, Thermo Fisher Scientific). Protein samples in Laemmli buffer + DTT were denatured at 94 °C for 3 min prior to running on SDS-PAGE gels (6% gels for co-IP and glycerol gradients, 15% gels for histone extracts, and 4–15% gradient gels for whole-cell protein lysates). Gels containing histone extracts were wet transferred to nitrocellulose membranes at 4 °C for 3 h at 400 mA current, then dried at room temperature followed by re-hydration in TBS + 0.1% Tween-20 (TBS-T) and blocking with Odyssey blocking buffer (LI-COR). All other gels were semi-dry transferred to PVDF using a Trans-Blot Turbo (Bio-Rad) according to the manufacturer’s protocol designed for high molecular weight proteins, and blocked with either 5% BSA or 5% milk in TBS. The following primary antibodies were used: anti-ARID1A (D2A8U, Cell Signaling), anti-CHD4 (D4B7, Cell Signaling), anti-ZMYND8 (A302-089, Bethyl), anti-ZMYND8 (Atlas), anti-BRG1 (ab110641, abcam), anti-BAF155 (D7F8S, Cell Signaling), anti-HDAC1 (10E2, Cell Signaling), anti-histone H3.3 (ab176840, abcam), anti-histone H3.3 (2D7-H1, abnova), and anti-histone H3 (D1H2, Cell Signaling). IRDye fluorescent dye (LI-COR) secondary antibodies were used for LI-COR fluorescence-based protein visualization of histones. Horseradish peroxidase (HRP) conjugated secondary antibodies (Cell Signaling) were used for chemiluminescence-based protein visualization of all other targets. Clarity Western ECL substrate (Bio-Rad) was used to activate HRP for chemiluminescence, captured by ChemiDoc XRS+ imaging system (Bio-Rad).
mRNA-seq and analysis
Seventy-two hours after initial siRNA transfection, and 24 h after low-sera conditioning, 12Z cells were purified for RNA using the Quick-RNA Miniprep Kit (Zymo Research). Transcriptome libraries (n = 3 replicates) were prepared and sequenced by the Van Andel Genomics Core from 500 ng of total RNA using the KAPA mRNA HyperPrep kit (v4.17) (Kapa Biosystems). RNA was sheared to 300–400 bp. Prior to PCR amplification, cDNA fragments were ligated to IDT for Illumina unique dual adapters (IDT DNA Inc). Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies), QuantiFluor dsDNA System (Promega), and Kapa Illumina Library Quantification qPCR assays (Kapa Biosystems). Individually indexed libraries were pooled, and 50 bp, paired-end sequencing was performed on an Illumina NovaSeq 6000 sequencer using a 100-cycle sequencing kit (Illumina). Each library was sequenced to an average raw depth of 20–25 million reads. Base calling was done by Illumina RTA3 and output of NCS was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.9.0.
For analysis, briefly, raw reads were trimmed with cutadapt [80] and Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) followed by quality control analysis via FastQC [81] and MultiQC [82]. Trimmed reads were aligned to hg38 assembly and indexed to GENCODE (v28) along with gene feature counting via STAR [83]. Low count genes with less than 1 count per sample on average were filtered prior to count normalization and differential gene expression (DGE) analysis by DESeq2 with empirical Bayes shrinkage for fold-change estimation [84, 85]. Wald probabilities were corrected for multiple testing by independent hypothesis weighting (IHW) [86] for downstream analyses. In presented analyses, “log2FC” is the empirically observed log2 fold-change in expression between conditions, while “slog2FC” is a moderated log2 fold-change estimate that removes noise from low count genes using the apeglm shrinkage estimator as implemented in DESeq2 [87]. Pairwise comparisons between different DGE analyses and gene sets were initially filtered for genes with transcripts commonly detected in both cell populations.
Histone peptide arrays
Anti-acetyl-H2A.Z (K4/K7) (D3V1I, Cell Signaling) antibody specificity was analyzed via histone peptide microarrays as previously described [88] with minor modifications. Arrays were designed in ArrayNinja [89] and printed using a 2470 Arrayer (Quanterix). All hybridization and wash steps were performed at ambient temperature. Slides were blocked with hybridization buffer (1× PBS [pH 7.6], 0.1% Tween, 5% BSA) for 30 min, then incubated with primary antibody diluted 1:1000 in hybridization buffer for 1 h. Slides were washed 3× for 5 min with PBS, then probed with Alexa647-conjugated secondary antibody diluted 1:5000 in hybridization buffer for 30 min. Slides were washed 3× for 5 min with PBS, dipped in 0.1× PBS to remove salt, and spun dry. Slides were scanned on an InnoScan 1100 microarray scanner (Innopsys), and images were analyzed and quantified using ArrayNinja. Plots were generated in Prism (GraphPad). Each peptide antigen is printed six times per array, and each antibody was screened on two separate arrays.
Chromatin immunoprecipitation (ChIP-seq) and analysis
Wild-type and lentiviral shRNA particle transduced 12Z cells were treated with 1% formaldehyde in growth media for 10 min at ambient temperature. Formaldehyde was quenched by the addition of 0.125 M Glycine and incubation for 5 min at room temperature, followed by PBS wash and scra**. 1×107 crosslinked cells were used for each ChIP, and each antibody and condition for ChIP was performed in duplicate. Chromatin from crosslinked cells was fractionated by digestion with micrococcal nuclease using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) according to the manufacturer protocol, followed by 30 s of sonication. ChIP was then performed according to the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling) with the addition of 5 mM sodium butyrate to preserve histone acetylation. To each 1.25 mL IP, the following antibodies were used: 1:125 anti-histone H3.3 (2D7-H1, abnova); 1:250 anti-histone H3.1/3.2 (61629, Active Motif); 1:50 anti-histone H2A.Z-acetyl (K4/K7) (D3V1I, Cell Signaling); 1:250 anti-histone H2A.Z (ab4174, abcam); 1:50 anti-acetyl-histone H4 (06-866, Millipore); 1:125 anti-histone H4K16ac (39167, Active Motif); 1:50 anti-CHD4 (D4B7, Cell Signaling); 1:250 anti-ZMYND8 (A302-089, Bethyl). Crosslinks were reversed with 0.4 mg/mL Proteinase K (Thermo Fisher) and 0.2 M NaCl at 65 °C for 2 h. DNA was purified using the ChIP DNA Clean & Concentrator Kit (Zymo).
Libraries for input (n = 1 per condition) and IP (n = 2) samples were prepared by the Van Andel Research Institute Genomics Core. Ten nanograms of material was used for input samples, and the entire precipitated sample was used for IPs. Libraries were generated using the KAPA Hyper Prep Kit (v5.16) (Kapa Biosystems). Prior to PCR amplification, end-repaired and A-tailed DNA fragments were ligated to IDT for Illumina UDI Adapters (IDT DNA Inc.). Quality and quantity of the finished libraries were assessed using a combination of Agilent DNA High Sensitivity chip (Agilent Technologies), QuantiFluor® dsDNA System (Promega), and Kapa Illumina Library Quantification qPCR assays (Kapa Biosystems). Individually indexed libraries were pooled, and 50 bp, paired-end sequencing (for ZMYND8, H3.3, H2A.Zac, and H4K16ac) or 100 bp, single-end sequencing (for CHD4, H2A.Z, and pan-H4ac) was performed on an Illumina NovaSeq 6000 sequencer using a 100-cycle sequencing kit (Illumina). Each library was sequenced to minimum read depth of 80 million reads per input library and 40 million reads per IP library. Base calling was performed by Illumina NCS v2.0, and NCS output was demultiplexed and converted to FastQ format with Illumina Bcl2fastq v1.9.0.
New and re-analyzed (differential) ChIP-seq experiments were analyzed as previously described [27]. Briefly, wild-type CHD4 and differential H2A.Z and pan-H4ac ChIP-seq experiments were analyzed as single-end libraries, while wild-type ZMYND8 and differential H3.3, H2A.Zac, and H4K16ac ChIP-seq were analyzed as paired-end libraries. Raw reads for IPs and inputs were trimmed with cutadapt [80] and Trim Galore! (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) followed by quality control analysis via FastQC [81] and MultiQC [82]. Trimmed reads were aligned to GRCh38.p12 reference genome [90] via Bowtie2 [91] with flag “--very-sensitive.” Aligned reads were sorted and indexed with samtools [92]. Only properly paired read fragments were retained for paired-end libraries via samtools view with flag “-f 3” followed by sorting and indexing. For libraries intended for differential analyses, molecular complexity was then estimated from duplicate rates by ATACseqQC [93] and preseqR [94], and libraries were subsampled to equivalent molecular complexity within an experimental design based on these estimates with samtools. Picard MarkDuplicates (http://broadinstitute.github.io/picard/) was used to remove PCR duplicates, followed by sorting and indexing. MACS2 [95] was used to call peaks on each ChIP replicate against the respective input control. For CHD4 and ZMYND8 IPs, MACS2 called broadPeaks with FDR < 0.05 threshold and otherwise default settings. For H2A.Z and H2A.Zac IPs, MACS2 called narrowPeaks with FDR < 0.05 threshold and flags “--nomodel --extsize 146” to bypass model building. For H3.3, pan-H4ac, and H4K16ac IPs, MACS2 called broadPeaks with FDR < 0.05 threshold and flags “--nomodel --extsize 146” to bypass model building. The resulting peaks were repeat-masked by ENCODE blacklist filtering and filtered for non-standard contigs [96]. A naive overlap** peak set, as defined by ENCODE [97], was constructed by calling peaks on pooled replicates followed by bedtools intersect [98] to select for peaks of at least 50% overlap with each biological replicate.
ChIP-seq differential histone abundance analysis (n = 2 per condition) was performed with csaw [99]. First, a consensus peak set was constructed for each differential experiment from the union of replicate-intersecting, filtered MACS2 peak regions called in each condition. When examining the effects of ARID1A knockdown on canonical H3.1/3.2 abundance at H3.3-marked sites, the H3.3 peak set was utilized for this analysis. ChIP reads were counted in these query regions by csaw, then filtered for low abundance peaks with average log2CPM < −3. When comparing ChIP libraries, any global differences in IP efficiency observed between the two conditions were considered a result of technical bias to ensure a highly conservative analysis. As such, we employed a loess-based local normalization to the peak count matrix, as is implemented in csaw [99], to assume a symmetrical MA distribution. A design matrix was then constructed from one “condition” variable. The count matrix and loess offsets were then supplied to edgeR [100] for estimating dispersions and fitting quasi-likelihood generalized linear models for differential abundance hypothesis testing. Nearby query regions were then merged up to 500 bp apart for a maximum merged region width of 5 kb, and the most significant probability was used to represent the merged region. Finally, FDR < 0.05 threshold was used to define significant differentially abundant regions.
Chromatin state modeling and optimization
The same genome-wide chromatin 18-state map of 12Z cells with or without ARID1A depletion, constructed with ChromHMM [37, 101] using total RNA, ATAC, H3K4me1, H3K4me3, H3K18ac, H3K27ac, and H3K27me3 data [27], was re-analyzed in Figs. 1, 2, and 5 studies. A refined ChromHMM model was constructed with further addition of H3.3, H2A.Z, H2A.Zac (K4/K7), pan-H4ac (K5/K8/K12/K16), and H4K16ac features with some procedural modifications. In order to reduce technical confounders in differential chromatin state analysis between control and ARID1A-depleted cell types, we adopted an equalized binarization framework described by Fiziev et al. [102]. Briefly, the ChromHMM chromosomal signal intermediate files during BAM binarization were saved and imported into R. Feature signal values were then background-subtracted by respective control signals when available (e.g., input chromatin for ChIP; does not occur for ATAC). For each feature and cell type, those (background-subtracted) signal values were ranked, and the top n ranked binarization calls are selected, where n is the lower number of calls among the two cell types for the given feature. The result is a new equalized binarization, where each feature has the same number of “present” region calls in both cell types, per chromosome. As an example, if H3K18ac called 27,000 present regions on chromosome 1 in control cells and 35,000 present regions in ARID1A-depleted cells, then the top 27,000 regions are retained in both cell types. Chromatin state models from 5 to 40 states were then computed using the “concatenated” approach to unify both cell types for differential state comparisons. The new chromatin state model was optimized at 25 states through a strategy devised by Gorkin et al. [103], which utilizes the ChromHMM CompareModels function to compare feature emission parameters from the 40-state (most complex) model against all other simpler models, as well as a k-means clustering of emission probabilities from all models together and analyzing the goodness of fit. See Additional file 1: Fig. S6 for related analyses. Across both strategies, 25 states was observed as a threshold for >95% median maximal state correlation and goodness of fit (between-cluster vs. total sum-of-squares) relative to the most complex model.
Bioinformatics and statistics
The human endometrioma vs. control endometrium genome-wide expression (Illumina BeadChips) data set [53] was retrieved from GEO accession GSE23339 and analyzed via GEO2R and limma [104,105,106]. biomaRt was used for all gene nomenclature and mouse-human ortholog conversions [107]. The cumulative hypergeometric distribution was calculated in R for enrichment tests. HOMER was used to quantify sequencing reads across sets of genomic regions including heatmaps [108]. GenomicRanges functions were used to intersect and manipulate genomic coordinates [109]. IGV [110] was used for visualizing epigenomic data across hg38 loci as MACS2 enrichment log-likelihood ratio (logLR) for ChIP-seq and ATAC-seq or FPKM for RNA-seq. Hierarchical clustering by Euclidean distance and heatmaps were generated by ComplexHeatmap [111]. ggplot2 was used for some plots in this study [112]. The statistical language R was used for various computing functions throughout this study [113].
Availability of data and materials
All data generated or analyzed during this study are included in this published article, its supplementary information files and publicly available repositories. The novel data sets supporting the conclusions of this article are available in the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) at SuperSeries accession GSE190557 [114]. Previously reported data were retrieved from GEO at accessions GSE121198 [115], GSE148474 [116], and GSE23339 [117] and re-analyzed as previously described [25, 27] or as stated in the “Methods” section.
Abbreviations
- ATAC:
-
Assay for Transposase-Accessible Chromatin
- ATAC-seq:
-
Assay for Transposase-Accessible Chromatin followed by sequencing
- ChIP:
-
Chromatin immunoprecipitation
- ChIP-seq:
-
Chromatin immunoprecipitation followed by sequencing
- DGE:
-
Differential gene expression
- EMT:
-
Epithelial-to-mesenchymal transition
- FDR:
-
False discovery rate
- FE:
-
Fold-enrichment
- GEO:
-
Gene Expression Omnibus
- Co-IP:
-
Co-immunoprecipitation
- log2FC:
-
log2 fold-change
- logLR:
-
log-likelihood ratio
- n.s.:
-
Not significant
- p :
-
Probability [of null hypothesis]
- PCA:
-
Principal component analysis
- OR:
-
Odds ratio
- SE:
-
Super-enhancer
- TSS:
-
Transcription start site
References
Whitehouse I, Flaus A, Cairns BR, White MF, Workman JL, Owen-Hughes T. Nucleosome mobilization catalysed by the yeast SWI/SNF complex. Nature. 1999;400(6746):784–7.
Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, et al. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol Cell. 2010;38(4):590–602.
Kassabov SR, Zhang B, Persinger J, Bartholomew B. SWI/SNF unwraps, slides, and rewraps the nucleosome. Mol Cell. 2003;11(2):391–403.
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
Clapier CR. Sophisticated conversations between chromatin and chromatin remodelers, and dissonances in cancer. Int J Mol Sci. 2021;22(11). https://pubmed.ncbi.nlm.nih.gov/34070411/.
Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, et al. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996;15(19):5370–82.
Alver BH, Kim KH, Lu P, Wang X, Manchester HE, Wang W, et al. The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun. 2017;8:14648.
Morris SA, Baek S, Sung MH, John S, Wiench M, Johnson TA, et al. Overlap** chromatin-remodeling systems collaborate genome wide at dynamic chromatin transitions. Nat Struct Mol Biol. 2014;21(1):73–81.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45(6):592–601.
Kadoch C, Crabtree GR. Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci Adv. 2015;1(5):e1500447.
Mittal P, Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol. 2020;17(7):435–48.
He S, Wu Z, Tian Y, Yu Z, Yu J, Wang X, et al. Structure of nucleosome-bound human BAF complex. Science. 2020;367(6480):875–81.
Mashtalir N, D'Avino AR, Michel BC, Luo J, Pan J, Otto JE, et al. Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 2018;175(5):1272–1288 e1220.
Dallas PB, Pacchione S, Wilsker D, Bowrin V, Kobayashi R, Moran E. The human SWI-SNF complex protein p270 is an ARID family member with non-sequence-specific DNA binding activity. Mol Cell Biol. 2000;20(9):3137–46.
Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol Cell Biol. 2013;33(2):265–80.
Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife. 2017;6. https://pubmed.ncbi.nlm.nih.gov/28967863/.
Barutcu AR, Lajoie BR, Fritz AJ, McCord RP, Nickerson JA, van Wijnen AJ, et al. SMARCA4 regulates gene expression and higher-order chromatin structure in proliferating mammary epithelial cells. Genome Res. 2016;26(9):1188–201.
Wu RC, Wang TL, Shih Ie M. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15(6):655–64.
Cancer Genome Atlas Research N, Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, et al. Integrated genomic characterization of endometrial carcinoma. Nature. 2013;497(7447):67–73.
Zondervan KT, Becker CM, Missmer SA. Endometriosis. N Engl J Med. 2020;382(13):1244–56.
Samartzis EP, Samartzis N, Noske A, Fedier A, Caduff R, Dedes KJ, et al. Loss of ARID1A/BAF250a-expression in endometriosis: a biomarker for risk of carcinogenic transformation? Mod Pathol. 2012;25(6):885–92.
Anglesio MS, Papadopoulos N, Ayhan A, Nazeran TM, Noe M, Horlings HM, et al. Cancer-associated mutations in endometriosis without cancer. N Engl J Med. 2017;376(19):1835–48.
Wiegand KC, Shah SP, Al-Agha OM, Zhao Y, Tse K, Zeng T, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N Engl J Med. 2010;363(16):1532–43.
Jones S, Wang TL, Shih Ie M, Mao TL, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330(6001):228–31.
Wilson MR, Reske JJ, Holladay J, Wilber GE, Rhodes M, Koeman J, et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat Commun. 2019;10(1):3554.
Reske JJ, Wilson MR, Holladay J, Wegener M, Adams M, Chandler RL. SWI/SNF inactivation in the endometrial epithelium leads to loss of epithelial integrity. Hum Mol Genet. 2020;29(20):3412–30.
Wilson MR, Reske JJ, Holladay J, Neupane S, Ngo J, Cuthrell N, et al. ARID1A mutations promote P300-dependent endometrial invasion through super-enhancer hyperacetylation. Cell Rep. 2020;33(6):108366.
Rafati H, Parra M, Hakre S, Moshkin Y, Verdin E, Mahmoudi T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011;9(11):e1001206.
Van Rechem C, Boulay G, Leprince D. HIC1 interacts with a specific subunit of SWI/SNF complexes, ARID1A/BAF250A. Biochem Biophys Res Commun. 2009;385(4):586–90.
Bui CB, Le HK, Vu DM, Truong KD, Nguyen NM, Ho MAN, et al. ARID1A-SIN3A drives retinoic acid-induced neuroblastoma differentiation by transcriptional repression of TERT. Mol Carcinog. 2019;58(11):1998–2007.
Shi L, Wen H, Shi X. The histone variant H3.3 in transcriptional regulation and human disease. J Mol Biol. 2017;429(13):1934–45.
Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res. 2011;21(3):421–34.
Chen P, Zhao J, Wang Y, Wang M, Long H, Liang D, et al. H3.3 actively marks enhancers and primes gene transcription via opening higher-ordered chromatin. Genes Dev. 2013;27(19):2109–24.
Deaton AM, Gomez-Rodriguez M, Mieczkowski J, Tolstorukov MY, Kundu S, Sadreyev RI, et al. Enhancer regions show high histone H3.3 turnover that changes during differentiation. Elife. 2016;5. https://pubmed.ncbi.nlm.nih.gov/27304074/.
Martire S, Gogate AA, Whitmill A, Tafessu A, Nguyen J, Teng YC, et al. Phosphorylation of histone H3.3 at serine 31 promotes p300 activity and enhancer acetylation. Nat Genet. 2019;51(6):941–6.
Zhang H, Gan H, Wang Z, Lee JH, Zhou H, Ordog T, et al. RPA interacts with HIRA and regulates H3.3 deposition at gene regulatory elements in mammalian cells. Mol Cell. 2017;65(2):272–84.
Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods. 2012;9(3):215–6.
Pillidge Z, Bray SJ. SWI/SNF chromatin remodeling controls Notch-responsive enhancer accessibility. EMBO Rep. 2019;20(5). https://pubmed.ncbi.nlm.nih.gov/30914409/.
Gehre M, Bunina D, Sidoli S, Lubke MJ, Diaz N, Trovato M, et al. Lysine 4 of histone H3.3 is required for embryonic stem cell differentiation, histone enrichment at regulatory regions and transcription accuracy. Nat Genet. 2020;52(3):273–82.
Cheneby J, Menetrier Z, Mestdagh M, Rosnet T, Douida A, Rhalloussi W, et al. ReMap 2020: a database of regulatory regions from an integrative analysis of human and Arabidopsis DNA-binding sequencing experiments. Nucleic Acids Res. 2020;48(D1):D180–8.
Savitsky P, Krojer T, Fujisawa T, Lambert JP, Picaud S, Wang CY, et al. Multivalent histone and DNA engagement by a PHD/BRD/PWWP triple reader cassette recruits ZMYND8 to K14ac-rich chromatin. Cell Rep. 2016;17(10):2724–37.
Guo R, Zheng L, Park JW, Lv R, Chen H, Jiao F, et al. BS69/ZMYND11 reads and connects histone H3.3 lysine 36 trimethylation-decorated chromatin to regulated pre-mRNA processing. Mol Cell. 2014;56(2):298–310.
Wen H, Li Y, ** Y, Jiang S, Stratton S, Peng D, et al. ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature. 2014;508(7495):263–8.
Adhikary S, Sanyal S, Basu M, Sengupta I, Sen S, Srivastava DK, et al. Selective recognition of H3.1K36 dimethylation/H4K16 acetylation facilitates the regulation of all-trans-retinoic acid (ATRA)-responsive genes by putative chromatin reader ZMYND8. J Biol Chem. 2016;291(6):2664–81.
Li N, Li Y, Lv J, Zheng X, Wen H, Shen H, et al. ZMYND8 reads the dual histone mark H3K4me1-H3K14ac to antagonize the expression of metastasis-linked genes. Mol Cell. 2016;63(3):470–84.
Gong F, Chiu LY, Cox B, Aymard F, Clouaire T, Leung JW, et al. Screen identifies bromodomain protein ZMYND8 in chromatin recognition of transcription-associated DNA damage that promotes homologous recombination. Genes Dev. 2015;29(2):197–211.
Spruijt CG, Luijsterburg MS, Menafra R, Lindeboom RG, Jansen PW, Edupuganti RR, et al. ZMYND8 co-localizes with NuRD on target genes and regulates poly(ADP-Ribose)-dependent recruitment of GATAD2A/NuRD to sites of DNA damage. Cell Rep. 2016;17(3):783–98.
Kraushaar DC, Chen Z, Tang Q, Cui K, Zhang J, Zhao K. The gene repressor complex NuRD interacts with the histone variant H3.3 at promoters of active genes. Genome Res. 2018;28(11):1646–55.
Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, et al. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell. 2016;165(2):331–42.
Serresi M, Kertalli S, Li L, Schmitt MJ, Dramaretska Y, Wierikx J, et al. Functional antagonism of chromatin modulators regulates epithelial-mesenchymal transition. Sci Adv. 2021;7(9). https://pubmed.ncbi.nlm.nih.gov/33627422/.
Colino-Sanguino Y, Cornett EM, Moulder D, Smith GC, Hrit J, Cordeiro-Spinetti E, et al. A read/write mechanism connects p300 bromodomain function to H2A.Z acetylation. iScience. 2019;21:773–88.
Rothbart SB, Krajewski K, Strahl BD, Fuchs SM. Peptide microarrays to interrogate the "histone code". Methods Enzymol. 2012;512:107–35.
Hawkins SM, Creighton CJ, Han DY, Zariff A, Anderson ML, Gunaratne PH, et al. Functional microRNA involved in endometriosis. Mol Endocrinol. 2011;25(5):821–32.
Grandi G, Toss A, Cortesi L, Botticelli L, Volpe A, Cagnacci A. The association between endometriomas and ovarian cancer: preventive effect of inhibiting ovulation and menstruation during reproductive life. Biomed Res Int. 2015;2015:751571.
Borrelli GM, Abrao MS, Taube ET, Darb-Esfahani S, Kohler C, Chiantera V, et al. (Partial) Loss of BAF250a (ARID1A) in rectovaginal deep-infiltrating endometriosis, endometriomas and involved pelvic sentinel lymph nodes. Mol Hum Reprod. 2016;22(5):329–37.
Konev AY, Tribus M, Park SY, Podhraski V, Lim CY, Emelyanov AV, et al. CHD1 motor protein is required for deposition of histone variant H3.3 into chromatin in vivo. Science. 2007;317(5841):1087–90.
Nakayama T, Shimojima T, Hirose S. The PBAP remodeling complex is required for histone H3.3 replacement at chromatin boundaries and for boundary functions. Development. 2012;139(24):4582–90.
Pradhan SK, Su T, Yen L, Jacquet K, Huang C, Cote J, et al. EP400 deposits H3.3 into promoters and enhancers during gene activation. Mol Cell. 2016;61(1):27–38.
Wang W, Vilella F, Alama P, Moreno I, Mignardi M, Isakova A, et al. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat Med. 2020;26(10):1644–53.
Zhang C, Wang X, Anaya Y, Parodi L, Cheng L, Anderson ML, et al. Distinct molecular pathways in ovarian endometrioid adenocarcinoma with concurrent endometriosis. Int J Cancer. 2018;143(10):2505–15.
Sikora J, Wroblewska-Czech A, Smycz-Kubanska M, Mielczarek-Palacz A, Cygal A, Witek A, et al. The role of complement components C1q, MBL and C1 inhibitor in pathogenesis of endometriosis. Arch Gynecol Obstet. 2018;297(6):1495–501.
Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44(12):1310–5.
Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33(4):690–705 e699.
Kovac K, Sauer A, Macinkovic I, Awe S, Finkernagel F, Hoffmeister H, et al. Tumour-associated missense mutations in the dMi-2 ATPase alters nucleosome remodelling properties in a mutation-specific manner. Nat Commun. 2018;9(1):2112.
Tong JK, Hassig CA, Schnitzler GR, Kingston RE, Schreiber SL. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature. 1998;395(6705):917–21.
Xue Y, Wong J, Moreno GT, Young MK, Cote J, Wang W. NURD, a novel complex with both ATP-dependent chromatin-remodeling and histone deacetylase activities. Mol Cell. 1998;2(6):851–61.
Basta J, Rauchman M. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res. 2015;165(1):36–47.
Ha M, Kraushaar DC, Zhao K. Genome-wide analysis of H3.3 dissociation reveals high nucleosome turnover at distal regulatory regions of embryonic stem cells. Epigenetics Chromatin. 2014;7(1):38.
Marques JG, Gryder BE, Pavlovic B, Chung Y, Ngo QA, Frommelt F, et al. NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. Elife. 2020;9. https://pubmed.ncbi.nlm.nih.gov/32744500/.
Jiao F, Li Z, He C, Xu W, Yang G, Liu T, et al. RACK7 recognizes H3.3G34R mutation to suppress expression of MHC class II complex components and their delivery pathway in pediatric glioblastoma. Sci Adv. 2020;6(29):eaba2113.
Gong F, Clouaire T, Aguirrebengoa M, Legube G, Miller KM. Histone demethylase KDM5A regulates the ZMYND8-NuRD chromatin remodeler to promote DNA repair. J Cell Biol. 2017;216(7):1959–74.
Ghosh K, Tang M, Kumari N, Nandy A, Basu S, Mall DP, et al. Positive regulation of transcription by human ZMYND8 through its association with P-TEFb complex. Cell Rep. 2018;24(8):2141–2154 e2146.
Trizzino M, Barbieri E, Petracovici A, Wu S, Welsh SA, Owens TA, et al. The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 2018;23(13):3933–45.
Bottardi S, Mavoungou L, Pak H, Daou S, Bourgoin V, Lakehal YA, et al. The IKAROS interaction with a complex including chromatin remodeling and transcription elongation activities is required for hematopoiesis. PLoS Genet. 2014;10(12):e1004827.
Schwabish MA, Struhl K. The Swi/Snf complex is important for histone eviction during transcriptional activation and RNA polymerase II elongation in vivo. Mol Cell Biol. 2007;27(20):6987–95.
Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, et al. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J. 2006;25(17):3986–97.
Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol. 2010;190(5):741–9.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–19.
Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of oncogenic super-enhancers is coupled with DNA repair by RAD51. Cell Rep. 2019;29(3):560–572 e564.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10–2.
FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–8.
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550.
Love MI, Anders S, Kim V, Huber W. RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000Res. 2015;4:1070.
Ignatiadis N, Klaus B, Zaugg JB, Huber W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat Methods. 2016;13(7):577–80.
Zhu A, Ibrahim JG, Love MI. Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics. 2019;35(12):2084–92.
Cornett EM, Dickson BM, Rothbart SB. Analysis of histone antibody specificity with peptide microarrays. J Vis Exp. 2017;126. https://pubmed.ncbi.nlm.nih.gov/28809825/.
Dickson BM, Cornett EM, Ramjan Z, Rothbart SB. ArrayNinja: an open source platform for unified planning and analysis of microarray experiments. Methods Enzymol. 2016;574:53–77.
Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen HC, Kitts PA, et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017;27(5):849–64.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Ou J, Liu H, Yu J, Kelliher MA, Castilla LH, Lawson ND, et al. ATACseqQC: a Bioconductor package for post-alignment quality assessment of ATAC-seq data. BMC Genomics. 2018;19(1):169.
Daley T, Smith AD. Predicting the molecular complexity of sequencing libraries. Nat Methods. 2013;10(4):325–7.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137.
Amemiya HM, Kundaje A, Boyle AP. The ENCODE Blacklist: identification of problematic regions of the genome. Sci Rep. 2019;9(1):9354.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–31.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Lun AT, Smyth GK. csaw: a Bioconductor package for differential binding analysis of ChIP-seq data using sliding windows. Nucleic Acids Res. 2016;44(5):e45.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40.
Ernst J, Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat Protoc. 2017;12(12):2478–92.
Fiziev P, Akdemir KC, Miller JP, Keung EZ, Samant NS, Sharma S, et al. Systematic epigenomic analysis reveals chromatin states associated with melanoma progression. Cell Rep. 2017;19(4):875–89.
Gorkin DU, Barozzi I, Zhao Y, Zhang Y, Huang H, Lee AY, et al. An atlas of dynamic chromatin landscapes in mouse fetal development. Nature. 2020;583(7818):744–51.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47.
Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics. 2007;23(14):1846–7.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, et al. NCBI GEO: archive for functional genomics data sets--update. Nucleic Acids Res. 2013;41(Database issue):D991–5.
Smedley D, Haider S, Ballester B, Holland R, London D, Thorisson G, et al. BioMart--biological queries made easy. BMC Genomics. 2009;10:22.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–89.
Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, et al. Software for computing and annotating genomic ranges. PLoS Comput Biol. 2013;9(8):e1003118.
Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–6.
Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32(18):2847–9.
Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer-Verlag; 2016.
R Core Team: R: a language and environment for statistical computing. 2018.
Reske JJ, Chandler RL. ARID1A-dependent maintenance of H3.3 is required for repressive CHD4-ZMYND8 chromatin interactions at super-enhancers. 2022, NCBI Gene Expression Omnibus, GSE190557. https://identifiers.org/geo:GSE190557. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE190557.
Wilson MR, Reske JJ, Chandler RL. ARID1A and PI3-Kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. 2019, NCBI Gene Expression Omnibus, GSE121198. https://identifiers.org/geo:GSE121198. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121198.
Wilson MR, Reske JJ, Chandler RL. ARID1A mutations promote P300-dependent endometrial invasion through super-enhancer hyperacetylation. 2020, NCBI Gene Expression Omnibus, GSE148474. https://identifiers.org/geo:GSE148474. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE148474.
Creighton C, Matzuk M, Hawkins S. Gene expression profiles of endometriosis. 2010, NCBI Gene Expression Omnibus, GSE23339. https://identifiers.org/geo:GSE23339. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23339.
Acknowledgements
We thank the Van Andel Research Institute Genomics Core for providing sequencing facilities and services. We thank Dr. Jeremy Prokop for the use of his ÄKTA start FPLC to prepare linear glycerol gradients.
Funding
RLC was supported by awards from the National Institute of Child Health and Human Development (R01HD103617 and R21HD099383) from NIH. MRW was supported by a National Cancer Institute Transition to Independence K99 Award (CA25152) from NIH. JH was supported by an F32 award (CA260116) from NIH. SBR was supported by an award from the National Institute of General Medical Sciences (GM124736) from NIH.
Author information
Authors and Affiliations
Contributions
JJR and RLC conceptualized the study. JJR generated, analyzed, and curated all new experimental genomic data, led integrative computational and statistical analysis, and performed bioinformatic investigation of public data resources. MRW performed the cell cycle analysis. BA, SH, and CP assisted with experiments. MA led the library construction and sequencing efforts. JH and SBR performed histone peptide array experiments for antibody characterization. RLC supervised the study and provided funding. JJR and RLC drafted the manuscript. JJR, MRW, BA, SH, CP, JH, MA, SBR, SAM, ATF, and RLC read, critiqued, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Information
. Figure S1. Supplemental ARID1A knockdown differential H3.3 data. Figure S2. H3.3 knockdown functional analysis. Figure S3. Effects of CHD4 loss on H3.3 chromatin and comparison to ARID1A. Figure S4. Additional chromatin features profiled in 12Z cells. Figure S5. Peptide specificity of anti-acetyl-H2A.Z (K4/K7). Figure S6. ChromHMM model optimization. Figure S7. Chromatin accessibility repressed by ARID1A is associated with H4 acetylation. Figure S8. Chromatin feature correlation across promoter-proximal super-enhancers. Figure S9. siCHD4/siZMYND8 functional analysis. Figure S10. H4K16ac enrichment at repressed mechanistic genes. Figure S11. Additive transcriptional repression by ARID1A and CHD4. Figure S12. Uncropped Western blots.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Reske, J.J., Wilson, M.R., Armistead, B. et al. ARID1A-dependent maintenance of H3.3 is required for repressive CHD4-ZMYND8 chromatin interactions at super-enhancers. BMC Biol 20, 209 (2022). https://doi.org/10.1186/s12915-022-01407-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-022-01407-y