
Abstract
Background
Karat (Musa troglodytarum L.) is an autotriploid Fe’i banana of the Australimusa section. Karat was domesticated independently in the Pacific region, and karat fruit are characterized by a pink sap, a deep yellow-orange flesh colour, and an abundance of β-carotene. Karat fruit showed non-climacteric behaviour, with an approximately 215-day bunch filling time. These features make karat a valuable genetic resource for studying the mechanisms underlying fruit development and ripening and carotenoid biosynthesis.
Results
Here, we report the genome of M. troglodytarum, which has a total length of 603 Mb and contains 37,577 predicted protein-coding genes. After divergence from the most recent common ancestors, M. troglodytarum (T genome) has experienced fusion of ancestral chromosomes 8 and 9 and multiple translocations and inversions, unlike the high synteny with few rearrangements found among M. schizocarpa (S genome), M. acuminata (A genome) and M. balbisiana (B genome). Genome microsynteny analysis showed that the triplication of MtSSUIIs due to chromosome rearrangement may lead to the accumulation of carotenoids and ABA in the fruit. The expression of duplicated MtCCD4s is repressed during ripening, leading to the accumulation of α-carotene, β-carotene and phytoene. Due to a long terminal repeat (LTR)-like fragment insertion upstream of MtERF11, karat cannot produce large amounts of ethylene but can produce ABA during ripening. These lead to non-climacteric behaviour and prolonged shelf-life, which contributes to an enrichment of carotenoids and riboflavin.
Conclusions
The high-quality genome of M. troglodytarum revealed the genomic basis of non-climacteric behaviour and enrichment of carotenoids, riboflavin, flavonoids and free galactose and provides valuable resources for further research on banana domestication and breeding and the improvement of nutritional and bioactive qualities.
Similar content being viewed by others


Background
Bananas (Musa spp.) are among the most favoured fruits worldwide and are important staple foods for people in some tropical and subtropical countries [1]. According to their distinct genetic backgrounds, four genomes are represented within Musa spp., including Musa acuminata (A genome, 2n = 2x = 22), Musa balbisiana (B genome, 2n = 2x = 22), Musa schizocarpa (S genome, 2n = 2x = 22) and the Australimusa species (T genome, 2n = 2x = 20) [2]. Bananas are typically triploids or diploids of subspecies of Musa acuminata or of Musa balbisiana and M. acuminata. Of the seven species of Australimusa, there is a distinct group of banana species known as Fe’i banana (Musa troglodytarum L.). Fe’i banana plants are characterized by erect bunches and produce fruit that have a deep yellow-orange flesh colour and an abundance of α-carotene and β-carotene [3]. Karat is a cultivar of Fe’i banana distributed on the island of Pohnpei. Karat used to be a traditional weaning food in Pohnpei and has regained popularity due to a campaign that promoted karat to combat vitamin A deficiency (VAD) [4]. Moreover, karat has also been found to be rich in riboflavin (vitamin B2), an essential vitamin for nervous system function and iron utilization [5]. Riboflavin also contributes to the yellow colour of karat flesh.
Carotenoids are a large group of isoprenoids that play essential roles in plants; carotenoids function as pigments in both photosynthesis and light harvesting and serve as substrates for the biosynthesis of strigolactone and abscisic acid (ABA) [6]. The carotenoid biosynthetic pathway has been described in plants [7]. 1-Deoxy-D-xylulose-5-phosphate synthase (DXS) is the first and rate-limiting enzyme of the MEP pathway, and phytoene synthase regulates the first step of carotenoid biosynthesis by condensation of geranylgeranyl diphosphate, which is rate-limiting [8, 9]. Both α- and β-carotene are produced through the cyclization of lycopene by ε-lycopene cyclase (LCYE) and β-lycopene cyclase (LCYB). The pro-vitamin A (PVA) content of Musa germplasms varied from 0 to 85.08 μg/g, with yellow-orange plantains, Papua New Guinea diploids and deep yellow-orange pulp Fe’i bananas have relatively high carotenoid contents [10]. The biosynthesis of carotenoids of the Fe’i cultivar Asupina has been examined, and MtCCD4 is thought to be the key gene that results in hyperaccumulation in Asupina [11]. MtPSY2a cloned from Asupina was successfully used to generate transgenic bananas with high PVA levels [12]. In Cavendish, MaSPL6 functions as an activator of MaLCYB1.1, and MaLCYB1.2 and plays essential roles in carotenoid accumulation during ripening [13]. High carbohydrate contents were also shown to contribute to the hyperaccumulation of carotenoids in the green mutant of the plantain variety Obubit Ntanga [14].
Fe’i banana fruit are a parthenocarpic edible type [15]. The domestication of Fe’i banana occurred independently of that of plantains and other banana species through parthenocarpy and sterility processes [15]. To date, the genomes of A, B, S and Musa itinerans of Musa spp. have been published [16,17,18,19]. In addition to the latest updated genome of Musa acuminata DH PaHang, the genome data of Musa acuminata Banksii, Zebrina, and Calcutta 4 were deposited in the banana genome hub [20]. A cross-genus pangenome of banana contains representatives of the Musa and Ensete genera was presented, including genomic short reads of Musa troglodytarum L. ‘Pisang Tongkat Langit’ (tongkat), a cultivar from Eastern Indonesia [21, 22]. However, limited information is available for the Musa troglodytarum L. (T) genome, which has restricted the mining and utilization of valuable germplasm and gene resources. In this study, we de novo sequenced the genome of Musa troglodytarum L. for the first time, by integrating Oxford Nanopore, PacBio, Illumina and Hi-C sequencing techniques. Karat fruit showed non-climacteric behaviour, with an approximately 215-day bunch filling time. Metabolomic analyses and transcriptome sequencing were carried out to determine candidate genes involved in non-climacteric behaviour and the enrichment of carotenoids and riboflavin. The data from this study will be valuable for further research on improving the nutritional and bioactive qualities of banana fruit, prolonging shelf-life and reducing postharvest crop losses.
Results
Assembly of the T genome
The chromosomes of karat were fluorescently stained, and the result showed that karat is an autotriploid cultivar with 30 chromosomes (Additional file 1: Fig. S1). According to the genome survey, the T genome is 606~655 Mb in size and has a heterozygosity rate ranging from 1.25% (tongkat, TT) to 1.55% (karat, TTT) (Additional file 1: Fig. S2 and Additional file 1: Table S1-2). For genome sequencing, we generated 42 Gb of Nanopore reads, 6.9 Gb of PacBio reads and 42 Gb of Illumina reads (Additional file 1: Table S3-5). Using NextDenovo and NextPolish, we obtained an assembly with a total length of 918 Mb and contig N50 of 4.9 Mb (Additional file 1: Table S6). After purging haplotigs (Additional file 1: Fig. S3), we obtained 603 Mb contigs (Additional file 1: Table S7), and with 110 Gb of Hi-C reads mapped, the contigs were arranged into 10 chromosomes (Fig. 1a and Additional file 1: Fig. S4). BUSCO analysis showed that 97.7% of the BUSCO genes were assembled (Additional file 1: Table S8). The transcriptomes of leaves, roots, stems and fruits were sequenced for gene annotation. Using Maker2 [23], we predicted 37,577 protein-coding genes (Additional file 1: Table S9). BUSCO analysis showed that 92.5% of the BUSCO genes were predicted (Additional file 1: Table S10). Using eggNOG-mapper [24], we predicted 30,377 protein-coding genes with orthologues, 16,687 genes with GO annotation and 13,105 genes with KEGG annotation. Using RepeatMasker [25], we found that 59.62% of the T genome contained repeat elements (Additional file 1: Table S11). LTR/Gypsy and LTR/Copia accounted for 15.1% and 36.4% of the genome, respectively. As specific marker of the centromeric regions in M. acuminata genome [16, 26], Nanica LINE clusters also presented in all chromosomes (Additional file 1: Fig. S5). Using LTR FINDER [27], we identified 3,128 intact LTRs, and analysis of the insertion time showed that there was an LTR insertion burst at 1.47 MYA (Additional file 1: Fig. S6), which occurred before the burst of M. balbisiana (0.32 MYA) and after the burst of M. acuminata (1.77 MYA). Then, Illumina reads of karat and tongkat were mapped to the T genome. There were 516,884 and 459,137 indels, and 7,716,375 and 7,125,857 single-nucleotide polymorphism (SNP) sites identified in karat and tongkat, respectively (Additional file 1: Table S12 and Additional file 1: Fig. S7).

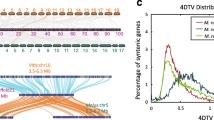
Overview of the T genome. a Chromosome overview of the T genome. A, GC content; B, repeat content; C LTR Copia content; D, LTR gypsy content; and E, gene content. b Erect fruit bunch of 25 DAF karat, which is pictured at Danzhou, Hainan, China. c Ancestor genome analysis and chromosome rearrangements. The bars representing the chromosomes of the A, B, S and T genomes are divided into 11 groups. Each colour presents one of the ancestral chromosomes. Phylogenetic analysis (d) and distribution of the 4 dTv distances between gene pairs (e) of the A, B, S and T genomes
Using OrthoFinder [28], we identified 8924 single-copy genes and 27,100 orthologous gene sets in the A, B, S, T and M. itinerans genomes. There were 7791 genes specific in the T genome. According to the phylogenetic tree generated by OrthoFinder and the divergence time of M. acuminata and M. balbisiana reported in a previous study [17], we constructed an ultrametric tree showing that M. troglodytarum diverged from the ancestor of M. acuminata, M. schizocarpa and M. balbisiana 20.8 MYA (Fig. 1d). According to the 4DTV distances, the peak Ks values were approximately 0.06, 0.06, and 0.07 for M. troglodytarum–M. acuminata, M. troglodytarum–M. schizocarpa and M. troglodytarum–M. balbisiana, respectively, where a peak Ks value of approximately 0.46 indicated whole-genome duplication (Fig. 1e). Using Café [29], we identified 771, 460, 1820 and 608 expanded gene families and 3325, 3049, 562 and 6980 contracted gene families in the A, S, T and B genomes, respectively. There are 11 gene families with rapid expansions in T genome. GO functional enrichment analysis was conducted to explore the functions T genome-specific genes and rapidly evolving families (Additional file 1: Fig. S8-9 and supplementary Additional file 2: Data 1-2). GO enrichment analysis show that those genes of rapidly evolving gene families were enriched in cell morphogenesis, cell growth, defense response to insect immune system process, defense response to bacterium, defense response to fungus, response to virus etc.
Using MGRA2 [30], we constructed the ancestral genome of the A, B, S and T genomes, which resulted in 11 contiguous ancestral regions (CARs) and 20,056 ordered ancestral genes. For chromosome rearrangement, we constructed bar plots of the A, B, S and T genomes compared to CARs using MCSCAN (Fig. 1c). The ancestor of the A, B, S and T genomes experienced multiple chromosome rearrangements before and after their divergence. Chromosome 1 of the A, B and T genomes experienced translocation after divergence, and chromosomes8 and 9 in the ancestry fused into chromosome 9 of the T genome. A dot plot of the synteny gene blocks between M. troglodytarum and M. acuminata also indicated the fusion of chromosomes 8 and 9 in the T genome (Additional file 1: Fig. S10-11). M. troglodytarum was domesticated independently and diverged from an ancestor of M. acuminata, M. schizocarpa and M. balbisiana 20.8 MYA. Thus, M. troglodytarum has experienced multiple translocations and inversions, unlike the high synteny with few rearrangements found among M. schizocarpa, M. acuminata and M. balbisiana.
Transcriptome and metabolome of the fruit
To determine the basis of the enrichment of carotenoids and flavonoids and the non-climacteric behaviour of karat, we integrated widely targeted metabolomics and targeted metabolomics data from karat pulp at 25, 45, 65, 115, 145, 173, 200 and 215 days after flowering (DAF) and RNA sequencing (RNA-seq) data from karat pulp at 25, 45, 65, 115, 100, 130, 145, 152, 159, 173, 200 and 208 DAF (Fig. 2a). According to widely targeted metabolomic data, we identified 877 metabolites, including flavonoids, lipids, phenolic acids, amino acids and their derivatives, organic acids, nucleotides and their derivatives, alkaloids, lignin, coumarins, tannins, terpenoids, quinones and others, 768 of them were divided into 5 clusters (Fig. 2f, Additional file 1: Fig. S12 and Additional file 2: Data 3-4). Cluster 2 represents the metabolites that increased during ripening, including alkaloids, amino acids and their derivatives, coumarins, free fatty acids, organic acids, phenolic acids, saccharides and alcohols, vitamins and others.

Targeted metabolomics and widely targeted metabolomics analysis of karat pulp at different developmental stages. a Transverse section of karat fruit at different developmental stages. Quantitation of carotenoids (b and c, μg/g) and relative quantification of flavonoids (c) and vitamin B (c). F1, (E/Z)-phytoene; F2, Lutein; F3, α-Carotene; F4, β-Carotene; B2, riboflavin; B5, D-pantothenic acid; B6, pyridoxine; F1, 4′-hydroxy-5,7-dimethoxyflavanone; F2, epicatechin; F3, myricetin-3-O-rutinoside; and F4, delphinidin-3-O-rutinoside. d Pink sap of pesudostem and fruits at 25 DAF. e Quantitation of flavonoids in pink sap. f Heatmap of metabolites in fruit pulp
Carotenoids were enriched throughout fruit development
According to the quantification of the karat pulp metabolites, lutein accumulated throughout all the fruit developmental stages, with a content of 10.62–32.25 μg/g (Fig. 2b, c). The contents of α-carotene, β-carotene and phytoene increased rapidly at 200 DAF and 215 DAF, with values of 53.42, 27.90, and 10.57 μg/g, respectively, at 215 DAF. In addition, β-cryptoxanthin-laurate, rubixanthin-laurate β-cryptoxanthin, γ-carotene and others, were also increased at 215 DAF (Additional file 2: Data 5). Microsynteny analysis of carotenoid biosynthesis pathway genes showed that MtSSUIIs were triplicated and MtCCD4s were duplicated in the T genome (Figs. 1c and 3b). According to the RNA-seq data, the key genes of carotenoid biosynthesis, including MtGGPPS1, MtSSUIIs, MtPSY2s, MtLCYBs, MtLCYEs, MtZDSs, Mtβ-OH and Mtε-OH, were all highly expressed across all the fruit developmental stages (Fig. 3a, c). At 200 DAF and 215 DAF, hyperaccumulation of α-carotene, β-carotene and phytoene coincided with a decrease in CCD4 expression.

Schematic representation of the carotenoid biosynthesis pathway and duplication of key enzymes in karat. Geranylgeranyl pyrophosphate synthase small subunit (SSU-II) and carotenoid cleavage dioxygenase 4 (CCD4) are duplicated in the T genome compared to the A, B, and S genomes. Phytoene, α-carotene and β-carotene contents were highly enriched in karat pulp. a Schematic representation of the vitamin E and carotenoid biosynthesis pathways in karat. Interactions are represented by red dotted lines. IPP, isopentenyl diphosphate; DMAPP, dimethylallyl diphosphate; GGPP, geranylgeranyl pyrophosphate; GGPPS, geranylgeranyl pyrophosphate synthase; PSY, phytoene synthase; PDS, phytoene desaturase; ZDS, ζ-carotene desaturase; LCYB, lycopene β-cyclase; LCYE, lycopene ε-cyclase; β-OH, carotene β-hydroxylase; ε-OH, ε-hydroxylase; ZEP, zeaxanthin epoxidase; and NCED, 9-cis-epoxycarotenoid dioxygenase. b Distribution of SSUll and CCD4 genes and the distribution and synteny between the T genome and A genome. c Heatmap of carotenoid biosynthesis pathway genes in different tissues
The triplication of MtSSUII may explain the enrichment of carotenoids in karat and other Fe’i banana fruits. The lutein contents of these fruits were high throughout the fruit development process. SSUII enhances the accumulation of carotenoids by interacting with GGPPS1 and PSY, promoting their enzymatic activity [31, 32]. In addition to MtSSUIIs enhancing the hyperaccumulation of carotenoids, MtCCD4, a key gene that regulates various branches of carotenoid biosynthesis, regulates the accumulation of α-carotene and β-carotene during ripening and is downregulated at the end of ripening [11]. Downregulation of CCD4 is fruit-specific and may be the key reason for the enrichment of only α-carotene and β-carotene in the fruit. According to coexpression network analysis, MtCCD4 was coregulated with MtETO1 and MtJAZ1. MtJAZ1 is the key regulator of the JA signalling pathway and is induced by JA [33]. Multiple JA response element G-box and TGACG-box motifs were identified in the promoters of MtCCD4a and MtCCD4b (Additional file 1: Fig. S13), similar to CCD4 in Brassica napus, indicating an extensive role of JA in the regulation of CCD4 [34]. In Osmanthus fragrans, OfCCD4 were also induced by JA treatment [35]. In the full-green (FG) stage, the decreased expression level of CCD4 coincides with the increase in the JA content, but in the full-ripening (FR) stage, the decrease in the JA content also coincides with the downregulation of MtCCD4s, implying the complex regulation of MtCCD4s by JA. For α-carotene and β-carotene rapidly accumulation in FR stage, JA may repress the accumulation of α-carotene and β-carotene by activating the expression of MtCCD4s in fruit. Therefore, further research is needed to elucidate the mechanism governing the regulation of MtCCD4s by JA, which may be spatiotemporally dependent and dose dependent.
Flavonoids are enriched early during fruit development, which may be due to the expansion of MtF3′5′Hs
The T genome has 17 F3′5 ′H loci, while there are eight, eight and five loci in the A, B and S genomes respectively. Microsynteny analysis showed that the flavonoid biosynthesis gene MtF3′5 ′H was tandemly duplicated on both chromosomes 2 and 10, resulting in nine more loci than were present in the A genome (Fig. 4b). In particular, F3′5′H on chromosome 9, which is a single locus in the A, B and S genomes, is duplicated into eight loci in the T genome. Moreover, seven of the eight loci distributed on chromosome 9 of the T genome showed highly similar expression patterns in karat. MtF3′H, which competes with MtF3′5′Hs for substrates, was largely decreased in karat. Microsynteny analysis also showed that there are three M. troglodytarum-specific regions in the upstream sequences of MtF3′H. No similar sequences were identified by BLAST in A, B, S or other genomes. The specific regions may contribute to the low expression level of MtF3’H in karat. According to the quantification of flavonoids in pink stem sap, the delphinidin-3-rutinoside chloride content was enriched. Moreover, multiple flavonoids in the pulp were found to be enriched, including 4′-hydroxy-5,7-dimethoxyflavanone, epicatechin, myricetin-3-O-rutinoside, and delphinidin-3-O-rutinoside. In particular, only 4′-hydroxy-5,7-dimethoxyflavanone was enriched at 215 DAF, while epicatechin, myricetin-3-O-rutinoside and delphinidin-3-O-rutinoside degraded largely at the end of the ripening process, coinciding with the fading of pink sap in the fruit (Fig. 2a, c–e). The duplication of MtF3′5′H and suppression of MtF3′H led to the enrichment of delphinidin-3-O-rutinoside, which differs from other types of bananas. The heatmap shows that the key genes involved in the synthesis of flavonoids were downregulated at the end of the ripening process, except for MtUFGTs, which were highly expressed throughout the ripening process (Fig. 4a, c).

Schematic representation of the flavonoid biosynthesis pathway and duplication of key enzymes in karat. a Schematic representation of the flavonoid biosynthesis pathway. PAL, phenylalanine ammonia-lyase; C4H, cinnamate-4-hydroxylase; 4CL, 4-coumarate-CoA ligase; CHS, chalcone synthase; CHI, chalcone isomerase; F3H, flavanone 3-hydroxylase; F3′H, flavonoid 3′-hydroxylase; F3′5′H, flavonoid 3′,5′-hydroxylase; DFR, dihydroflavonol 4-reductase; ANS, leucoanthocyanidin dioxygenase; ANR, anthocyanidin reductase; LAR, leucoanthocyanidin reductase; FNS, flavone synthase; FLS, flavonol synthase; UFGT, anthocyanidin 3-O-glucosyltransferase; RT, UDP-rhamnose; and MT, anthocyanin O-methyltransferase. b Distribution of duplicated F3′5′Hs in the T and A genomes. c Heatmap of flavonoid biosynthesis pathway genes in different tissues. d Characteristic of the 5′ UTR upstream of F3′H
Riboflavin is enriched in karat pulp
According to the widely targeted metabolome analysis, riboflavin (B2), pantothenic acid (B5) and pyridoxine (B6) were enriched in karat pulp (Fig. 2c). In particular, riboflavin (B2) was enriched, especially in Fe’i banana fruit. Transcriptome analysis also showed that the riboflavin de novo synthesis genes MtRIBA1 and MtFMNse showed higher expression levels in karat fruit pulp than in BXJ (Ba** melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genomics. 2015;16(1):1–20." href="#ref-CR51" id="ref-link-section-d261401456e1897_2">51,52,53]. Banana is usually a climacteric fruit, while karat shows non-climacteric behaviour with the transformation of ethylene-induced ripening into ABA-induced ripening. The triplication of MtSSUII resulted in the enhancement of ABA biosynthesis, and the suppression of ethylene biosynthesis by a mutation in MtERF11 may be the primary reason for the non-climacteric behaviour of karat. In banana, a complex network regulating fruit ripening has been reported, and a dual-loop circuit, involving NAC and MADS transcription factors and ethylene signalling, was shown to control banana fruit ripening [54]. MaMADS1, MaMADS2 and MaMADS36 are also necessary for banana fruit ripening [55,56,57]. However, the expression patterns of MtMADS1, MtMDAS2a and MtMADS2b, which are homologues MaMADS1 and MaMADS2, were different from those in BXJ; thus, these genes may function in an ethylene-dependent mode. A multilayered regulatory cascade comprising MaNAC1, MaNAC2, MaXB3, MaERF11, MaACS1 and MaACO1 is involved in ripening, of which MaERF11 functions as a repressor of ethylene biosynthesis by repressing MaACS1 and MaACO1 [39, 58, 59]. MaERF11 was repressed by MaNAC2 and MaNAC1; both genes were repressed by MaXB3. During ripening, the repression of MaACS1 was removed with the downregulation of MaXB3. However, in karat, the regulatory cascade was disrupted by the insertion of LTRs, and MtERF11 repressed the expression of MtACS1 throughout the fruit development process. Additionally, the comparative transcriptome analysis indicated that the regulatory network comprising NAC1, NAC2, XB3, ERF11, ACS1 and ACO1 was conserved in Fj, BXJ and karat. Moreover, ABA-related transactors such as ABI5, ASR1, and ASR2 were also involved in the regulation of fruit ripening in Fj, BXJ and karat. These data will provide important resources for further research on the fruit ripening process of banana.
Conclusions
In this study, we de novo sequenced the genome of Musa troglodytarum L. for the first time. Genome microsynteny analysis showed that the duplication of MtSSUIIs due to chromosome rearrangement may lead to the accumulation of carotenoids and ABA in the fruit. And the expression of duplicated MtCCD4s is repressed during ripening, leading to the accumulation of α-carotene, β-carotene and phytoene. The duplication of MtF3′5′H and suppression of MtF3′H led to the enrichment of delphinidin-3-O-rutinoside, which is different from that in the other types of bananas. The free galactose content in the pulp was higher than that in the pulp of other banana fruit and may be due to the sequence variations in the 5’ UTR and sixth intron of MtGALK. Due to an LTR fragment insertion upstream of MtERF11, karat cannot produce large amounts of ethylene but can produce ABA during ripening, resulting in non-climacteric behaviour and prolonging bunch filling time. In conclusion, integrating metabolome, comparative transcriptome and microsynteny analysis, we revealed the genomic basis of non-climacteric behaviour and enrichment of carotenoids, riboflavin, flavonoids, free galactose of karat (Musa troglodytarum L.). These data provide insights into the improvement of the nutritional and bioactive qualities, prolongation of shelf-life and reducing postharvest crop loss.
Methods
Sampling, sequencing and assembly
Karat plants growing in a greenhouse of the National Gene Bank of Tropical Crops in Danzhou, Hainan, China, were sampled for genome sequencing. This Musa germplasm was collected from Kosrae of The Federated States of Micronesia during a plant resources investigation and cooperation. The genomic DNA of leaves was extracted for genomic library construction. The DNA high-molecular-weight genomic DNA extraction was performed using an SDS-based method [60]. For Illumina sequencing, libraries with 350 bp insertions were constructed. For PacBio sequencing, libraries with 20,000 bp insertions were constructed and sequenced on the PacBio RS II system using P6-C4 chemistry. For Nanopore single-molecule sequencing, libraries with high-molecular-weight genomic DNA were constructed on PromethION. In total, 42,304,446,276 bp reads were produced by Nanopore single-molecule sequencing, 42,215,455,800 bp Illumina short reads were produced, and 6,961,206,933 bp PacBio reads were produced.
Hi-C libraries were created from young leaves of M. troglodytarum at BioMarker Technologies Company as described previously [61]. Briefly, the leaves and formaldehyde were mixed together and then lysed, and then the cross-linked DNA was digested with DpnII overnight. Sticky ends were biotinylated and proximity-ligated to form chimaeric junctions that were enriched and then physically sheared to a size of 500–700 bp. Chimaeric fragments representing the original cross-linked long-distance physical interactions were then processed into paired-end sequencing libraries, and 110 Gb of paired-end reads were produced on the Illumina HiSeq X Ten platform.
For RNA-seq, total RNA was extracted from leaves, roots, pseudostems and fruits using TRIzol reagent according to the manufacturer’s instructions. After removing genomic DNA using DNase I (Takara), mRNAs were obtained using oligo (dT) beads and subsequently broken into short fragments, followed by cDNA synthesis. Paired-end sequencing was conducted on a HiSeq X Ten platform (Illumina, CA, USA).
Genome assembly and annotation
Using GenomeScope 2.0 [62], a program that employs a polyploid-aware mixture model to assess heterozygous and polyploid genomes, we estimated the genome size using jellyfish-produced K-mer counts. Nanopore long reads were imported for assembly by NextDenovo v.2.3.0 (https://github.com/Nextomics/NextDenovo), with the errors first corrected by the NextCorrect module and then assembled into 918,212,466 bp contigs by the NextGraph module. Utilizing Illumina short reads and PacBio reads, NextPolish (https://github.com/Nextomics/NextPolish) was subsequently used to polish the preliminary assembly with the default parameters. Then, the allelic haplotigs were eliminated by Purge_Haplotigs (V1.1.1) [63]. The final assembly was then assembled into a scaffold based on proximity-guided assembly by ALLHIC [64].
RepeatModeler v1.0.11 with default parameters was used to identify TEs de novo [65]; this program employs two different software programs: RECON (v1.08) and RepeatScout (v1.0.5). For consensus building and classification steps, the consensus TE libraries generated above were imported into MAKER2 for further repeat annotations by employing RepeatMasker (v4.05). TEclass (v2.1.3) was subsequently used to classify unknown TEs using a support vector machine (SVM) method. Tandem Repeat Finder (v4.07) was then used to identify repeats within the genomes with the following parameters: ‘1 1 2 80 5 200 2,000 –d –h’.
To detect LTRs, the LTR_retriever pipeline with default parameters was used to integrate and remove false positives from the initial predictions of LTR FINDER [27, 66]. BUSCO v5.22 was used to evaluate genome completeness using the viridiplantae_odb10 dataset, which includes 425 single-copy and conserved protein-coding genes [67]. The LTR insertion time was estimated by the transcripts implemented in the LTR package using the formula T = K/2μ, with μ representing the neutral mutation rate and set to 1.38 × 10−8.
The repeats and protein-coding genes in the genome were annotated by MAKER2 (v3.01.02) [23]. The MAKER2 pipeline was processed twice to obtain high-quality gene annotations. The RNA-seq reads of the leaves, stems, roots and fruits were imported to Trinity to generate genome-guided and de novo assemblies, with the default parameters used. Then, the assemblies were imported into the PASA pipeline (v2.3.3) to construct a comprehensive transcript database. The comprehensive transcripts were used to train the predictors embedded in the MAKER2 pipeline, including AUGUSTUS (v3.3.1), GENEMARK (v3.5.2) and SNAP (version 2006-07-28). After filtering out proteins produced by MAKER2 that had low AED values, the ab initio predictors AUGUSTUS, GENEMARK and SNAP were trained again. Then, utilizing high-confidence and high-sensitivity transcripts produced by StringTie (v2.1.1) [68] as inputs, the MAKER2 pipeline was run again. Using eggNOG-mapper (v2) [24] and its default database, genes were assigned functional annotations.
Genome structure and evolution
The genome data of M. acuminata (GCA_000313855.2) [69], M. balbisiana (GCA_004837865.1) [70] and M. itinerans (GCA_001649415.1) [71] was downloaded from NCBI. The genome data of M. schizocarpa was downloaded at http://www.genoscope.cns.fr/plants [72]. The RNA-seq data of BXJ and FJ were downloaded from BioProject accession number PRJNA394594. The short reads genome sequencing data included in SRR15675960 for tongkat and SRR16526594 for karat were used to estimate genome size.
Orthologues were identified by OrthoFinder (v2.2.7) [28] with default parameters, using the longest transcripts of protein-coding genes from M. troglodytarum, M. acuminata, M. balbisiana, M. schizocarpa and M. itinerans. According to the orthologue set mentioned above and the results from MCScanX (v0.8) [73], putative protogenes (pPGs) were predicted. Then, the pPGs ordered according to the gene location in each species were integrated into GRIMM format and imported into MGRA2 [30] for ancestral genome reconstruction, in which heuristic higher breakpoint reuse was used. Then, the exhaustive set of ordered protogenes (oPGs) was imported for collinear genes reported by MCScanX [73]. The rooted tree generated by OrthoFinder was used for the construction of ultrametric trees of M. troglodytarum, M. acuminata, M. balbisiana and M. schizocarpa by r8s (v1.81) [74] with default parameters. Using Café (v4.2.1) [29] with default parmameters, we subsequently estimated the gene family expansion and contraction events among M. troglodytarum, M. acuminata, M. balbisiana and M. schizocarpa. Using R packages clusterProfiler [75] with parameters “pvalueCutoff = 0.05, pAdjustMethod = ‘BH’”, we performed GO enrichment of M. troglodytarum specific genes and rapidly evolving families.
Metabolomic analyses
Carotenoid, flavonoid, monosaccharide and disaccharide contents were analysed via targeted metabolomics. Karat pulp at 215 DAF was used for monosaccharide and disaccharide analysis. The pink sap of karat was used for flavonoid and carotenoid analysis. Karat pulp at 25, 45, 65, 115, 145, 173, 200 and 215 DAF was used for carotenoid analysis. The carotenoid, flavonoid, monosaccharide and disaccharide contents were analysed as described in previous studies [76,77,78]. The sample extracts were analysed using a UPLC-ESI-MS/MS system (ExionLC™ AD https://sciex.com.cn/; MS, Applied Biosystems 6500 Triple Quadrupole, https://sciex.com.cn/). Linear ion trap (LIT) and triple quadrupole (QQQ) scans were acquired on a QQQ-linear ion trap mass spectrometer (QTRAP), QTRAP® 6500+ LC–MS/MS system equipped with an ESI turbo ion-spray interface operating in positive ion mode and controlled by Analyst v1.6.3 software (AB-SCIEX).
For widely targeted metabolomics analyses, karat pulp at 25, 45, 65, 115, 145, 173, 200 and 215 DAF was used. The metabolomics analyses were performed as described in a previous study [79]. Briefly, the sample extracts were analysed using a UPLC–ESI–MS/MS system (Shimadzu Nexera X2, https://www.shimadzu.com.cn/; MS, Applied Biosystems 4500 QTRAP, https://www.thermofisher.cn/cn/zh/home/brands/applied-biosystems.html). LIT and QQQ scans were acquired on a triple quadrupole-linear ion trap mass spectrometer (QTRAP), AB4500 QTRAP UPLC/MS-MS system was equipped with an ESI turbo ion-spray interface operating in positive and negative ion mode and controlled by Analyst 1.6.3 software (AB-SCIEX).
Availability of data and materials
The Musa troglodytarum L. genome sequences and raw sequence data from RNA-seq with accessions SRX12729179-SRX1272927 and genome assembly with assession ASM2354706v1 have been deposited under BioProject accession number PRJNA772907 [80]. The GFF format Gene and TE annotation file of the Musa troglodytarum L. genome was available in figshare [81]. The cloned promoter sequences of MtACS1, MtACO1 and cDNA sequences of MtGALK and MtSSUIIs were deposited in figshare [82].
References
De Langhe E, Vrydaghs L, De Maret P, Perrier X, Denham T. Why bananas matter: an introduction to the history of banana domestication. Ethnobot Res Appl. 2009;7:165–77.
Davey MW, Gudimella R, Harikrishna JA, Sin LW, Khalid N, Keulemans J. A draft Musa balbisiana genome sequence for molecular genetics in polyploid, inter-and intra-specific Musa hybrids. BMC Genomics. 2013;14(1):1–20.
Englberger L. A community and laboratory-based assessment of natural food sources of vitamin A in the Federated States of Micronesia. Nutrition -- Micronesia (Federated States); 2003.
Englberger L, Schierle J, Marks GC, Fitzgerald MH. Micronesian banana, taro, and other foods: newly recognized sources of provitamin A and other carotenoids. J Food Compos Anal. 2003;16(1):3–19.
Mahabadi N, Bhusal A, Banks SW. Riboflavin Deficiency. StatPearls. 2020.
Saini RK, Keum Y-S. Significance of genetic, environmental, and pre-and postharvest factors affecting carotenoid contents in crops: a review. J Agric Food Chem. 2018;66(21):5310–24.
Sun T, Yuan H, Cao H, Yazdani M, Tadmor Y, Li L. Carotenoid metabolism in plants: the role of plastids. Mol Plant. 2018;11(1):58–74.
Fray RG, Wallace A, Fraser PD, Valero D, Hedden P, Bramley PM, et al. Constitutive expression of a fruit phytoene synthase gene in transgenic tomatoes causes dwarfism by redirecting metabolites from the gibberellin pathway. Plant J. 1995;8(5):693–701.
Ye X, Al-Babili S, Kloti A, Zhang J, Lucca P, Beyer P, et al. Engineering the provitamin A (β-carotene) biosynthetic pathway into (carotenoid-free) rice endosperm. Science. 2000;287(5451):303–5.
Amah D, Van Biljon A, Brown A, Perkins-Veazie P, Swennen R, Labuschagne M. Recent advances in banana (musa spp.) biofortification to alleviate vitamin A deficiency. Crit Rev Food Sci Nutr. 2019;59(21):3498-510.
Buah S, Mlalazi B, Khanna H, Dale JL, Mortimer CL. The quest for golden bananas: investigating carotenoid regulation in a Fe’i group Musa cultivar. J Agric Food Chem. 2016;64(16):3176–85.
Paul JY, Khanna H, Kleidon J, Hoang P, Geijskes J, Daniells J, et al. Golden bananas in the field: elevated fruit pro-vitamin A from the expression of a single banana transgene. Plant Biotechnol J. 2017;15(4):520–32.
Zhu L-s, Liang S-m, Chen L-l, Wu C-j, Wei W, Shan W, et al. Banana MaSPL16 modulates carotenoid biosynthesis during fruit ripening through activating the transcription of lycopene β-cyclase genes. J Agric Food Chem. 2019;68(5):1286–96.
Heng Z, Sheng O, Huang W, Zhang S, Fernie AR, Motorykin I, et al. Integrated proteomic and metabolomic analysis suggests high rates of glycolysis are likely required to support high carotenoid accumulation in banana pulp. Food Chem. 2019;297:125016.
Daniells J, Daniells J, Daniells J. Diversity in the genus Musa; 2001.
D’hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, et al. The banana (Musa acuminata) genome and the evolution of monocotyledonous plants. Nature. 2012;488(7410):213–7.
Wang Z, Miao H, Liu J, Xu B, Yao X, Xu C, et al. Musa balbisiana genome reveals subgenome evolution and functional divergence. Nat Plants. 2019;5(8):810–21.
Belser C, Istace B, Denis E, Dubarry M, Baurens F-C, Falentin C, et al. Chromosome-scale assemblies of plant genomes using nanopore long reads and optical maps. Nat Plants. 2018;4(11):879–87.
Wu W, Yang Y-L, He W-M, Rouard M, Li W-M, Xu M, et al. Whole genome sequencing of a banana wild relative Musa itinerans provides insights into lineage-specific diversification of the Musa genus. Sci Rep. 2016;6(1):1–11.
Droc G, Lariviere D, Guignon V, Yahiaoui N, This D, Garsmeur O, et al. The banana genome hub. Database. 2013;2013:bat035.
Ploetz RC, Kepler AK, Daniells J, Nelson SC. Banana and plantain—an overview with emphasis on Pacific island cultivars. Species Profiles Pac Island Agroforest. 2007;1:21–32.
Rijzaani H, Bayer PE, Rouard M, Doležel J, Batley J, Edwards D. The pangenome of banana highlights differences between genera and genomes. Plant Genome. 2022;15(1):e20100.
Cantarel BL, Korf I, Robb SM, Parra G, Ross E, Moore B, et al. MAKER: an easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 2008;18(1):188–96.
Cantalapiedra CP, Ana HP, Ivica L, Peer B, Jaime HC. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol Biol Evol. 2021;38(12):5825-9.
Tarailo-Graovac M, Chen N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr Protoc Bioinformatics. 2004;5(1):4-10.
Belser C, Baurens F-C, Noel B, Martin G, Cruaud C, Istace B, et al. Telomere-to-telomere gapless chromosomes of banana using nanopore sequencing. Commun Biol. 2021;4(1):1–12.
Ou S, Jiang N. LTR_FINDER_parallel: parallelization of LTR_FINDER enabling rapid identification of long terminal repeat retrotransposons. Mobile. DNA. 2019;10(1):1-3.
Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16(1):1–14.
Han MV, Thomas GWC, Jose LM, Hahn MW. Estimating Gene Gain and Loss Rates in the Presence of Error in Genome Assembly and Annotation Using CAFE3. Mol Biol Evol. 2013;30(8):1987–97.
Avdeyev P, Jiang S, Aganezov S, Hu F, Alekseyev MA. Reconstruction of ancestral genomes in presence of gene gain and loss. J Comput Biol. 2016;23(3):150–64.
Zhou F, Wang CY, Gutensohn M, Jiang L, Zhang P. A recruiting protein of geranylgeranyl diphosphate synthase controls metabolic flux toward chlorophyll biosynthesis in rice. Proceedings of the National Academy of Sciences. 2017;114(26):6866–71.
Wang Q, Huang XQ, Cao TJ, Zhuang Z, Wang R, Lu S. Heteromeric Geranylgeranyl Diphosphate Synthase Contributes to Carotenoid Biosynthesis in Ripening Fruits of Red Pepper (Capsicum annuum var. conoides). Journal of agricultural and food chemistry. 2018;66(44):11691–700.
Thines B, Katsir L, Melotto M, Niu Y, Mandaokar A, Liu G, et al. JAZ repressor proteins are targets of the SCF(COI1) complex during jasmonate signalling. Nature. 2007;448(7154):661.
Zhou XT, Jia LD, Duan MZ, Chen X, Li JN. Genome-wide identification and expression profiling of the carotenoid cleavage dioxygenase (CCD) gene family in Brassica napus L. PLoS One. 2020;15(9):e0238179.
Han Y, Wu M, Cao L, Yuan W, Dong M, Wang X, et al. Characterization of OfWRKY3, a transcription factor that positively regulates the carotenoid cleavage dioxygenase gene OfCCD4 in Osmanthus fragrans. Plant Mol Biol. 2016;91(4):485–96.
** L, Bing Y, Yangdong G. The role of abscisic acid in fruit ripening and responses to abiotic stress. J Exp Bot. 2013;65(16):4577–88.
Su L, Diretto G, Purgatto E, Danoun S, Zouine M, Li Z, et al. Carotenoid accumulation during tomato fruit ripening is modulated by the auxin-ethylene balance. BMC Plant Biol. 2015;15(1):1–12.
Yuan H, Zhang J, Nageswaran D, Li L. Carotenoid metabolism and regulation in horticultural crops. Horticult Res. 2015;2:15036.
Shan W, Kuang J-f, Wei W, Fan Z-q, Deng W, Li Z-g, et al. MaXB3 modulates MaNAC2, MaACS1, and MaACO1 stability to repress ethylene biosynthesis during banana fruit ripening. Plant Physiol. 2020;184(2):1153–71.
Lefebvre V, North H, Frey A, Sotta B, Seo M, Okamoto M, et al. Functional analysis of Arabidopsis NCED6 and NCED9 genes indicates that ABA synthesized in the endosperm is involved in the induction of seed dormancy. Plant J :Cell Mol Biol. 2006;45(3):309–19.
Jia H, Jiu S, Zhang C, Wang C, Tariq P, Liu Z, et al. Abscisic acid and sucrose regulate tomato and strawberry fruit ripening through the abscisic acid-stress-ripening transcription factor. Plant Biotechnol J. 2016;14(10):2045-65.
Phillips KM, McGinty RC, Couture G, Pehrsson PR, McKillop K, Fukagawa NK. Dietary fiber, starch, and sugars in bananas at different stages of ripeness in the retail market. PLoS One. 2021;16(7):e0253366.
Egert A, Peters S, Guyot C, Stieger B, Keller F. An Arabidopsis T-DNA insertion mutant for galactokinase (AtGALK, At3g06580) hyperaccumulates free galactose and is insensitive to exogenous galactose. Plant Cell Physiol. 2012;53(5):921–9.
Ferreira JP, Overton KW, Wang CL. Tuning gene expression with synthetic upstream open reading frames. Proc Natl Acad Sci U S A. 2014;110(28):11284–9.
Farcuh BS, Rivero RM, Shlizerman S, et al. Sugar metabolism reprogramming in a non-climacteric bud mutant of a climacteric plum fruit during development on the tree. J Exp Bot. 2017;68(21-22):5813–28.
Janssens SB, Vandelook F, De Langhe E, Verstraete B, Smets E, Vandenhouwe I, et al. Evolutionary dynamics and biogeography of M usaceae reveal a correlation between the diversification of the banana family and the geological and climatic history of Southeast Asia. New Phytol. 2016;210(4):1453–65.
Fu N, Ji M, Rouard M, Yan H-F, Ge X-J. Comparative plastome analysis of Musaceae and new insights into phylogenetic relationships. BMC Genomics. 2022;23(1):1–20.
Christelová P, Valárik M, Hřibová E, De Langhe E, Doležel J. A multi gene sequence-based phylogeny of the Musaceae (banana) family. BMC Evol Biol. 2011;11(1):1–13.
Villavicencio LE, Blankenship SM, Sanders DC, Swallow WH. Ethylene and carbon dioxide concentrations in attached fruits of pepper cultivars during ripening. Sci Hortic. 2001;91(1):17–24.
Fernández-Trujillo JP, Obando-Ulloa JM, Martínez JA, Moreno E, García-Mas J, Monforte AJ. Climacteric and non-climacteric behavior in melon fruit: 2. Linking climacteric pattern and main postharvest disorders and decay in a set of near-isogenic lines. Postharvest Biol Technol. 2008;50(2-3):125–34.
Saladié M, Cañizares J, Phillips MA, Rodriguez-Concepcion M, Larrigaudière C, Gibon Y, et al. Comparative transcriptional profiling analysis of develo** melon (Cucumis melo L.) fruit from climacteric and non-climacteric varieties. BMC Genomics. 2015;16(1):1–20.
Yamane M, Abe D, Yasui S, Yokotani N, Kimata W, Ushijima K, et al. Differential expression of ethylene biosynthetic genes in climacteric and non-climacteric Chinese pear fruit. Postharvest Biol Technol. 2007;44(3):220–7.
Fernandez i Marti A, Saski CA, Manganaris GA, Gasic K, Crisosto CH. Genomic sequencing of Japanese plum (Prunus salicina Lindl.) mutants provides a new model for Rosaceae fruit ripening studies. Front Plant Sci. 2018;9:21.
Lü P, Yu S, Zhu N, Chen YR, Zhou B, Pan Y, et al. Genome encode analyses reveal the basis of convergent evolution of fleshy fruit ripening. Nat Plants. 2018;4(10):784–91.
Yakir E, Zhangjun F, Sela N, Xu Y, Singh V, Dagar A, et al. MaMADS2 repression in banana fruits modifies hormone synthesis and signalling pathways prior to climacteric stage. Bmc. Plant Biol. 2018;18(1):1-17.
Elitzur T, Yakir E, Quansah L, Zhangjun F, Vrebalov JT, Khayat E, et al. Banana MaMADS Transcription Factors Are Necessary for Fruit Ripening and Molecular Tools to Promote Shelf-Life and Food Security. Plant Physiol. 2016;171(1):380-91.
Liu J, Liu M, Wang J, Zhang J, Miao H, Wang Z, et al. Transcription factor MaMADS36 plays a central role in regulating banana fruit ripening. J Exp Bot. 2021;72(20):7078–91.
Yun-Yi X, Jian-Ye C, Jiang-Fei K, Wei S, Hui X, Yue-Ming J, et al. Banana ethylene response factors are involved in fruit ripening through their interactions with ethylene biosynthesis genes. J Exp Bot. 2013;8:2499–510.
Han Y-C, Kuang J-F, Chen J-Y, Liu X-C, **ao Y-Y, Fu C-C, et al. Banana transcription factor MaERF11 recruits histone deacetylase MaHDA1 and represses the expression of MaACO1 and expansins during fruit ripening. Plant Physiol. 2016;171(2):1070–84.
**a Y, Chen FS, Du Y, Liu C, Bu G, **n Y, et al. A modified SDS-based DNA extraction method for raw soybean. Biosci Rep. 2019;39(2):BSR20182271.
Xu S, **ao S, Zhu S, Zeng X, **g L, Liu J, et al. A draft genome assembly of the Chinese sillago (Sillago sinica), the first reference genome for Sillaginidae fishes. Gigaence. 2018;9:9.
Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 2017;33(14):2202–4.
Roach MJ, Schmidt SA, Borneman AR. Purge Haplotigs: allelic contig reassignment for third-gen diploid genome assemblies. BMC Bioinformatics. 2018;19(1):1–10.
Zhang X, Zhang S, Zhao Q, Ming R, Tang H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants. 2019;5(8):833–45.
Flynn JM, Hubley R, Rosen J, Clark AG, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci. 2020;117(17):9451–7.
Ou S, Jiang N. LTR_retriever: A highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176(2):1410–22.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2.
Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, Salzberg SL. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. 2015;33(3):290–5.
D’hont A, Denoeud F, Aury J-M, Baurens F-C, Carreel F, Garsmeur O, et al. Musa acuminata subsp. malaccensis, genome sequencing and assembly. NCBI accession: GCA_000313855.2. (2012). https://www.ncbi.nlm.nih.gov/assembly/GCF_000313855.2.
Wang Z, Miao H, Liu J, Xu B, Yao X, Xu C, et al. Musa balbisiana, genome sequencing and assembly. NCBI accession: GCA_004837865.1. (2019). https://www.ncbi.nlm.nih.gov/assembly/GCA_004837865.1.
Wu W, Yang Y-L, He W-M, Rouard M, Li W-M, Xu M, et al. Musa itinerans, genome sequencing and assembly. NCBI accession: GCA_001649415.1. (2016). https://www.ncbi.nlm.nih.gov/assembly/GCA_001649415.1.
Belser C, Istace B, Denis E, Dubarry M, Baurens F-C, Falentin C, et al. Musa schizocarpa, genome sequencing and assembly. (2018). https://www.genoscope.cns.fr/externe/plants/chromosomes.html.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49-e.
Sanderson MJ. r8s: Inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19(2):301–2.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7.
Zhao C, Zayed O, Zeng F, Liu C, Zhang L, Zhu P, et al. Arabinose biosynthesis is critical for salt stress tolerance in Arabidopsis. New Phytol. 2019;224(1):274–90.
Zhou K, Hu L, Li Y, Chen X, Ma F. MdUGT88F1-mediated phloridzin biosynthesis regulates apple development and Valsa canker resistance. Plant Physiol. 2019;180(4):00494.2019.
**ong C, Luo D, Lin A, Zhang C, Shan L, He P, et al. A tomato B-box protein SlBBX20 modulates carotenoid biosynthesis by directly activating PHYTOENE SYNTHASE1, and is targeted for 26S proteasome-mediated degradation. New Phytol. 2019;221(1):279–94.
Zhou S, Kremling KA. Metabolome-Scale Genome-Wide Association Studies Reveal Chemical Diversity and Genetic Control of Maize Specialized Metabolites. The Plant Cell. 2019;31(5):937–55.
Jiabin w. Musa troglodytarum L., genome sequencing and assembly. NCBI accession: PRJNA772907. (2022). https://www.ncbi.nlm.nih.gov/bioproject/PRJNA772907.
Jiabin W. Gene and TE annotation file of the Musa troglodytarum L. genome. figshare; 2022. https://doi.org/10.6084/m9.figshare.20363118.v1.
Jiabin W. The cloned promoter sequences of MtACS1, MtACO1 and cDNA sequences of MtGALK and MtSSUIIs. figshare; 2022. https://doi.org/10.6084/m9.figshare.19767559.v1.
Acknowledgements
We thank Bart Panis at Bioversity International for his suggestions for this research and revising the manuscript.
Funding
This work is financially supported by the National Key Research and Development Plan Program (No. 2021YFC2600603), Science and Technology Cooperation Project of Hainan Key R&D Program (No. ZDYF2019214) and Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (No. 1630032019039).
Author information
Authors and Affiliations
Contributions
L.X. and Z.Y.L. designed the research; Z.Y.L., J.B.W., B.L.H., Y.L.F., Y.L.J., Y.C. and X.B.W. performed the experiments; L.X., Z.Y.L. and J.B.W. analysed the data; Q.Q.Y, Q.L.W. and C.Y.M. supplied the materials; J.B.W. and Z.Y.L. wrote the manuscript; and L.X. revised the manuscript. All authors critically read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1
. Fluorescent staining of karat chromosomes. The root tip of Musa troglodytarum L. karat plants staining with DPAI and screened under ultraviolet and fluorescence microscopy. Figure S2. GenomeScope profile of karat and tongkat. Figure S3. Bimodal histogram for Purge Haplotigs processing. The cutoff values for low, mid, and high points were 6,42 and 105, respectively. Figure S4. Hi-C map** of chromosomes of the T genome. Figure S5. Heatmap of density of Nanica LINE. Figure S6. Analysis of LTR insertion times of the A, B, S and T genomes. Ma, Musa acuminata; Mb, Musa balbisiana; Ms, Musa schizocarpa; and Mt, Musa troglodytarum L. Figure S7. Distribution of SNP and indel sites in karat and tongkat. Indel sites in tongkat (A) and karat (B) and SNP sites in tongkat (C) and karat (D). Figure S8. GO enrichment analysis of T genome specific genes. Figure S9. GO enrichment analysis rapidly evolving gene families in T genome. Figure S10. Dot plot of syntenic bocks between M. troglodytarum and M. acuminata. Figure S11. Synteny map of chromosome 8 and 9 among M. troglodytarum, M. acuminata and M. balbisiana. Figure S12. Clusters of metabolites in karat fruit pulp. DAF, days after flowering. Figure S13. The distribution of JA response element G-box and TGACG-box motifs in the promoters of MtCCD4s. Figure S14. The distribution of GCC-boxes in the promoters of MtACO1 and MtACS1. Figure S15. Expression patterns of genes involved in karat fruit ripening. Gene expression was normalized to FPKM (fragments per kilobase of transcript per million read pairs). DAF, days after flowering. Figure S16. Expression patterns of genes involved in FJ fruit ripening. Gene expression was normalized to FPKM (fragments per kilobase of transcript per million read pairs). FJ (Fen jiao), dwarf banana. DAF, days after flowering. DPH, days post-harvest. Figure S17. Expression patterns of genes involved in BXJ fruit ripening. Gene expression was normalized to FPKM (fragments per kilobase of transcript per million read pairs). BXJ (Ba** Jiao), Cavendish banana. Figure S18. Expression patterns of genes involved in starch degradation and galactose accumulation. The gene IDs of M. troglodytarum, M. acuminata and M. balbisiana start with ‘Mt’, ‘LOC’ and ‘THU’, respectively. Gene expression was normalized to FPKM (fragments per kilobase of transcript per million read pairs). Figure S19. The effect of exogenous application free galactose on shoot buds of karat and BXJ. The shoot buds of karat (a) and BXJ (b) grown 20 days under a controlled environment (10 h light, 35% relative humidity and 25 °C). The karat (c)and BXJ (d) shoot buds (2-3 cm) were transferred into half-strength MS supplemented with Gal (10 mM, B2; and 100 mM, B1) and sucrose (10 mM, Z1), respectively. Table S1. Summary of GenomeScope profile on Tongkat with a k-mer of 19. Table S2. Summary of GenomeScope profile on karat with a k-mer of 19. Table S3. summary of short reads of genome sequencing. Table S4. summary of Pacbio reads of genome sequencing. Table S5. Summary of Nanopore reads of genome sequencing. Table S6. Summary of BUSCO analysis of contigs before Purge Haplotigs processing (C:98.4%). Table S7. Summary contigs after Purge Haplotigs processing and correction of chimeric contigs using ALLHIC_correcter. Table S8. Summary of BUSCO analysis of genome (C:97.7%). Table S9. Summary of protein-coding genes of M. troglodytarum. Table S10. Summary of BUSCO analysis of predicted gene (C:92.5%). Table S11. Summary of Repeat content of genome. Table S12. Summary of SNP and Indel sites of Karat and Tongkat.
Additional file 2: Data 1
. List of M. troglodytarum specific genes. Genes were annotated by eggNOG-mapper. Data 2. List of genes of rapidly evolving families in M. troglodytarum. Genes were annotated by eggNOG-mapper. Data 3. Metabolites of karat pulp identified by widely targeted metabolomics analysis. Data 4. Clustered metabolites of karat pulp identified by widely targeted metabolomics analysis. Data 5. Carotenoid contents of pulp at different developmental stages. Data 6. Expression levels of genes involved in fruit ripening in karat. Data 7. Expression levels of genes involved in fruit ripening in BXJ. Data 8. Expression levels of genes involved in fruit ripening in FJ. Data 9. Sugar contents in the pulp of ripening karat fruit. Data 10. Expression levels of nodes of the ripening-related coexpression network.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Wang, J., Fu, Y. et al. The Musa troglodytarum L. genome provides insights into the mechanism of non-climacteric behaviour and enrichment of carotenoids. BMC Biol 20, 186 (2022). https://doi.org/10.1186/s12915-022-01391-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-022-01391-3