
Abstract
Background
Next generation sequencing (NGS) has been a handy tool in clinical practice, mainly due to its efficiency and cost-effectiveness. It has been widely used in genetic diagnosis of several inherited diseases, and, in clinical oncology, it may enhance the discovery of new susceptibility genes and enable individualized care of cancer patients. In this context, we explored a pan-cancer panel in the investigation of germline variants in Brazilian patients presenting clinical criteria for hereditary cancer syndromes or familial history.
Methods
Seventy-one individuals diagnosed or with familial history of hereditary cancer syndromes were submitted to custom pan-cancer panel including 16 high and moderate penetrance genes previously associated with hereditary cancer syndromes (APC, BRCA1, BRCA2, CDH1, CDKN2A, CHEK2, MSH2, MSH6, MUTYH, PTEN, RB1, RET, TP53, VHL, XPA and XPC). All pathogenic variants were validated by Sanger sequencing.
Results
We identified a total of eight pathogenic variants among 12 of 71 individuals (16.9%). Among the mutation-positive subjects, 50% were diagnosed with breast cancer and had mutations in BRCA1, CDH1 and MUTYH. Notably, 33.3% were individuals diagnosed with polyposis or who had family cases and harbored pathogenic mutations in APC and MUTYH. The remaining individuals (16.7%) were gastric cancer patients with pathogenic variants in CDH1 and MSH2. Overall, 54 (76.05%) individuals presented at least one variant uncertain significance (VUS), totalizing 81 VUS. Of these, seven were predicted to have disease-causing potential.
Conclusion
Overall, analysis of all these genes in NGS-panel allowed the identification not only of pathogenic variants related to hereditary cancer syndromes but also of some VUS that need further clinical and molecular investigations. The results obtained in this study had a significant impact on patients and their relatives since it allowed genetic counselling and personalized management decisions.
Similar content being viewed by others


Background
Hereditary cancer syndrome is a genetic predisposition to several types of cancer caused by pathogenic germline variants in one or more genes [1]. It corresponds to 5–10% of all cancers and have peculiar clinical aspects, such as the onset at an early age, high lifetime risk for multiple primary tumors, and cancer occurring in successive generations of the family [2, 3].
Most hereditary cancer syndromes display autosomal dominant inheritance involving genes that are mainly controlling cell cycle or DNA repair [4]. For example, mutations in BRCA1 and BRCA2 confer 40–80% lifetime risk of develo** breast cancer, and 11–50% of develo** ovarian cancer [5]; germline variants in at least one of the DNA mismatch repair genes (eg. MSH2, MLH1, MSH6, and PMS1) affect in up to 80% of the Lynch syndrome individuals [6]; and mutations in CDH1 gene are detected in 30 to 40% of families with hereditary diffuse gastric cancer (HDGC) [7].
For a long time, single-gene analyses were used for detection of the genetic cause of hereditary cancers. However, the recent advances in DNA sequencing techniques have allowed the application of next generation sequencing (NGS) in clinical practice, including in genetic diagnosis of inheritance disease. In addition to its rapid, efficient and cost-effective approach for testing multiple cancer susceptibility genes, it may enhance the discovery of new susceptibility genes and enable individualized care of cancer patients [8, 9].
Most genomic cancer studies are carried out in North America and Western Europe, which do not fully represent the admixed Brazilian genetic background [8]. Currently, Brazilian heritability of cancer risk has been widely explored, so further studies might allow the development of more reliable surveillance approaches and medical guidelines for subjects carrying pathogenic germline mutations. In this context, we built an NGS custom pan-cancer panel containing 16 high and moderate penetrance genes previously associated with hereditary cancer syndromes (APC, BRCA1, BRCA2, CDH1, CDKN2A, CHEK2, MSH2, MSH6, MUTYH, PTEN, RB1, RET, TP53, VHL, XPA and XPC) [10, 11]. Here, we report the application of this panel in the investigation of such germline variants in patients from Northern Brazil presenting clinical criteria for hereditary cancer syndromes or familial history.
Methods
Participants
This study included 71 individuals diagnosed or with familial history of hereditary cancer syndromes (hereditary breast and ovarian cancer – HBOC, hereditary diffuse gastric cancer, Lynch syndrome, familial adenomatous polyposis or MUTYH-associated polyposis), according to National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. They were recruited from João de Barros Barreto University Hospital (HUJBB, Belém, Pará, Brazil). The study was approved by the Institutional Review Board from HUJBB (CAAE: 89363618.3.0000.5634), and all participants gave their written informed consent. The patients were selected according to the clinical criteria for hereditary syndrome according to National Comprehensive Cancer Network (NCCN) Clinical Practice Guidelines in Oncology. Individuals who did not meet the clinical criteria for hereditary syndromes were excluded.
Next-generation sequencing
A custom pan-cancer panel was built containing 16 genes related to hereditary cancer syndromes (APC, BRCA1, BRCA2, CDH1, CDKN2A, CHEK2, MSH2, MSH6, MUTYH, PTEN, RB1, RET, TP53, VHL, XPA and XPC) [10]. Genomic DNA from the participants were extracted from whole blood samples collected during medical interview. DNA samples were quantified using Qubit 2.0 Fluorometer (Thermo Fisher Scientific), normalized to 25 ng/μL and custom pan-cancer library preparation. Briefly, indexed paired-end libraries were synthesized using TruSeq Custom Amplicon Library Prep Kit v1.5 (Illumina) and sequenced on MiSeq sequencing systems (Illumina). The raw sequencing reads of all libraries were deposited at the European Nucleotide Archive [Accession number: PRJEB43823].
Raw sequencing data were first filtered to remove low-quality reads and contaminants using Trimmomatic v.0.36 [12]. Resulting reads were aligned to the human genome (hg19) (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/) using BWA (v.0.7, [13]), and then sorted and indexed using sambamba [14] and samtools [15]. Small variants were identified using Genome Analysis Tool Kit (GATK) v3.8–0 [16] as follows. Alignments were recalibrated using RealignerTargetCreator and IndelRealigner tools against Mills & 1000G Gold Standard Indels (GATK resource bundle) (https://console.cloud.google.com/storage/browser/gcp-public-data%2D%2Dbroad-references/hg19/v0) [17]. Bases were recalibrated using BaseRecalibrator and dbSNP Build 138 (https://www.ncbi.nlm.nih.gov/snp/) [18] and variants were called using HaplotypeCaller. Joint genoty** was then performed using GenotypeGVCFs and the resulting SNPs and INDELs were filtered with VariantFiltration (SNPFilter with filterExpression: “QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0”; and INDELFilter with filterExpression: “QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0”). Finally, all variants were annotated with information from the dbNSFP database v3.5a (https://sites.google.com/site/jpopgen/dbNSFP) [19] using the snpEff v4.3 tool [20].
ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) was used to determine the clinical significance of all reported variants, in which variants were classified as pathogenic if they had a truncating effect. Pathogenicity of variants of uncertain significance (VUS) was investigated using ten different predictors: CADD [21], FATHMM [22], LRT [23], MetaSVM [24], MutationAssessor [25], MutationTaster [26], PROVEAN [27], Polyphen2 (HDIV and HVAR) [28] and SIFT [29], and those predicted by at least five predictors as likely pathogenic were considered to have a disease-causing potential. Variants were named according to Human Genome Variation Society nomenclature (HGVS, hpp://www.hgvs.org).
Pathogenic variants validation
Pathogenic variants presence were validated by Sanger sequencing in ABI 3130 (Applied Biosystems) as follows. The location of interest was amplified by PCR using primers shown in Supplementary Table S1. Sanger Sequencing was carried out with 1 μL of purified PCR product of each exon, 0.5 μL of the forward/reverse specific primer, 0.5 μL of ABI Prism Bid Dye Terminator Cycle Sequencing v3.1 Kit (Applied Biosystems, USA), 3 μL of SaveMoney buffer, and 10 μL of water to a final volume of 15 μL. The thermocycling reaction proceeded as follows: 96 °C for 1 min, followed by 35 cycles of 96 °C for 15 s, 50 °C for 15 s and 60 °C for 4 min. The sequence information was interpreted by ABI Analysis Software™. The electropherograms were analyzed using the Chromas 2.6.6 software and compared with the reference sequence obtained from NCBI.
Results
Pan-cancer panel
We analyzed data from 71 individuals, including 60 cancer patients - breast cancer (68.3%), gastric cancer (16.7%), and other types of cancer (15%) - and 11 cancer-free individuals with family history of cancer, mainly familial adenomatous polyposis (FAP) (81.8%) (Table 1).
A total of eight pathogenic (either pathogenic or likely pathogenic) variants were identified in APC, BRCA1, CDH1, MSH2 and MUTYH among 12 mutation-positive individuals (16.9%). Insertions and deletions represented 50% (n = 4) of all pathogenic variants, whereas single nucleotides variants (SNVs) accounted for the other half. The functional consequences of the identified pathogenic variants were primarily frameshift effects (50%), followed by stop gained (37.5%) and missense (12.5%) (Table 2), all of them were confirmed by Sanger sequencing. No variants described as pathogenic were identified in 11 genes (BRCA2, CDKN2A, CHEK2, MSH6, PTEN, RB1, RET, TP53, VHL, XPA and XPC).
Most of these pathogenic mutations identified were nonrecurrent (62.5%). Among the recurrent mutations, MUTYH c.1187 G > A (p.Gly396Asp) was reported in unrelated individuals, whereas BRCA1 c.1961delA (p.Lys654fs) and APC c.2195dupA (p.Asn732fs) were reported in related individuals.
Almost all probands with positive results for pathogenic variants had only a single mutation (n = 11), with the exception of one proband affected with colonic polyps who was compound heterozygous for MUTYH (c.1187G > A and c.1147delC) in accordance with the recessive pattern of MUTYH-associated polyposis.
Among the individuals with pathogenic variants, 50% were diagnosed with breast cancer and had mutations in BRCA1, CDH1 and MUTYH – gene not typically associated with this cancer. Notably, 33.3% were individuals diagnosed with polyposis or who had family cases and harbored pathogenic mutations in APC and MUTYH. The remaining individuals (16.7%) were gastric cancer patients with pathogenic variants in CDH1 and MSH2.
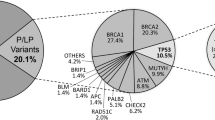
A total of 81 VUS were identified in APC, BRCA1, CDH1, CDKN2A, CHEK2, MSH2, MSH6, MUTYH, PTEN, RB1, RET, VHL and XPA. PTEN presented the highest amount of VUS with 22 variants (Fig. 1). Among all individuals tested, 54 (76.05%) presented at least one VUS and 14 individuals had a negative result for both pathogenic variants and VUS.

Count of variants of uncertain significance (VUS) by gene
Most VUS were in intronic, intragenic or untranslated regions (UTR) and, therefore, it was not possible to predict their pathogenicity (Supplementary Table S2). Only 12 variants were submitted to the prediction (Supplementary Table S3), among these seven were predicted to be deleterious by at least five predictors (six missense variants and one structural interaction variant), suggesting their disease-causing potential.
Family screening
The pan-cancer panel allowed us to identify familial mutation in affected patients, directing screening on family members and, consequently, to provide a personalized genetic counselling and management decisions. Examples of families that were benefited from the pan-cancer panel results are depicted in Fig. 2 and Fig. 3.

Pedigree of mutation-positive proband of family A. Black arrow indicates the proband with colonic polyps affected by two variants in MUTYH gene (c.1147delC and c.1187G > A) (III8). Red arrow indicates family members tested for mutations. A small red square indicates individuals carrying the mutation c.1147delC. A small blue square indicates individuals carrying the mutation c.1187G > A

Pedigree of mutation-positive proband of family B. Black arrow indicates the proband carrying BRCA1 c.1961delA mutation (II6). Small red square indicates individuals diagnosed with breast cancer. Small blue square indicates individuals diagnosed with bowel cancer. (+) represents individuals tested for mutation with BRCA1 c.1961delA mutation. (−) represents individuals tested for mutation without BRCA1 c.1961delA mutation
Family A (Fig. 2) proband was submitted to genetic testing due to colonic polyposis diagnosis. Two pathogenic variants in MUTYH gene were identified, c.1147delC and c.1187G > A. Nine unaffected family members were investigated – of these, three men and two women presented c.1147delC, two women presented c.1187G > A, while one man was compound heterozygous for these variants. Family B (Fig. 3) proband was submitted to genetic testing since they met the clinical criteria for HBOC. This individual presented BRCA1 c.1961delA. Sixteen family members were tested to this variant, being seven mutation-positive (four men and three women).
Therefore, as pathogenic variants were identified in unaffected individuals with an affected family member, it allowed the choosing of an appropriate surveillance approach and management decisions for the mutation-negative subjects.
Discussion
Advances in NGS technologies and their cost-effectiveness have made multigene panels a useful diagnostic tool in oncology clinic, particularly for offering an increase in mutation detection rate, which benefits individuals without family history information or atypical phenotype [30]. In the present study, a custom pan-cancer panel was developed and applied for the detection of hereditary cancer-related pathogenic mutations.
Variant analysis revealed the presence of at least one pathogenic variant in 16.9% of the analyzed individuals. Most of the individuals were breast cancer patients, being BRCA1 mutations responsible for 50% of the pathogenic variants in these patients. In the example of a family which was benefited from the multigene panel analysis, BRCA1 c.1961delA was also reported in men, demonstrating the importance of BRCA1 screening not only in women, since men with pathogenic variants in this gene have a slightly higher risk for prostate cancer and male breast cancer [31, 32].
Germline mutations in BRCA1 and BRCA2 are very relevant to the development of HBOC, however, they are responsible for only 15–25% of such cases, which demonstrates the significant contribution of other genes [33, 34]. In this study, the remaining pathogenic variants in breast cancer occurred in CDH1 and MUTYH.
CDH1 encodes E-cadherin, a protein responsible for calcium-dependent cell-to-cell adhesion. Germline mutations in this gene have been reported to cause hereditary diffuse gastric cancer (HDGC), a disorder that leads to an increased risk for diffuse gastric cancer and lobular breast cancer (LBC) [35]. However, substantial evidences have demonstrated an increased risk for LBC among CDH1 mutations carriers regarding their familial history for diffuse gastric cancer (DGC) [36]. In this study, CDH1 c.1003 C > T was reported in a breast cancer patient without familial history for DGC.
MUTYH mutations are the cause of MUTYH-associated polyposis (MAP), a syndrome that predisposes to colorectal polyposis and colorectal cancer [37]. Here, MUTYH c.1187G > A was reported in two unrelated individuals with breast cancer. This variant is the most frequent of all MUTYH mutations in various populations [38], but the association between this variant and breast cancer remains controversial. In Northern Israel, monoallelic inheritance was associated with an elevated risk of breast cancer [39], while in Non-Hispanic individuals of European ancestry there was no positive association [40]. Thus, our preliminary finding in the Brazilian population reinforces the need for further studies and for genetic counseling for families that have this variant segregating.
Here, one individual with colonic polyposis showed compound heterozygous mutations in MUTYH (c.1147delC and c.1187G > A). Genetic counselling was essential to identify other biallelic carriers in the family, in addition to monoallelic carriers (potential increased breast cancer risk), since these individuals may have a 2-fold increased risk for colorectal cancer and other cancers when compared to the general population [41].
Among the individuals with gastric cancer, one presented a mutation in CDH1 and, another, in MSH2. CDH1 alterations underlie HDGC, conferring risk of 56–70% higher [42]. Gastric cancer occurrence has a frequency of 1–6% in individuals with a Lynch syndrome-associated mutation, and this risk increases by 9% for those that present germline MSH2 mutations [43, 44]. In this study, a mutation in this gene was identified in a gastric cancer patient (c.388_389delCA) – until now, this variant had been reported only in Brazilian Lynch syndrome patients [45, 46].
Multigene pan-cancer panels that include a variety of cancer types might contribute to better understanding an individual’s risk for cancer, but it also raises new challenges in genetic counselling [9]. These challenges are even more complex in populations with a broad lack of information, such as the Brazilian one. Our population has several particular genomic features resulting from a high degree of admixture [47]. Most of the available genomic databases contain data from ancestry populations that do not entirely represent our genetic composition. It may explain the low number of pathogenic variants found in this study − it is possible that among the VUS and the “not described” variants there are some pathogenic variants that have not been associated with clinical data yet due to the lack of studies and validation [48]. Among the 81 VUS identified, seven were predicted with deleterious potential by at least five different predictors, reinforcing the need for functional studies that validate their pathogenicity.
Moreover, it is important to comprehend that most studies on cancer genetics were carried out in North America and Western Europe, thus reflecting the mutation burden in subjects of European ancestry. It is known that people of distinct ethnicities inherit a different pattern of pathogenic mutations from their ancestors. For instance, BRCA1 and BRCA2 show significant global variations according to contribution in regional cancer incidence and to mutation spectrum [49]. Only a minor part of the heritability of cancer risk has been elucidated so far, and further whole exome sequencing studies are needed to significantly increase the identification of hereditary cancer genes [8].
Despite the small sample size, which may not fully represent the Brazilian population, our results suggest the existence of a unique genetic background which needs to be more explored. Considering Brazil as a continental country submitted to different colonization processes, further studies should include samples from the different regions of the country.
Conclusions
In conclusion, our findings contributes to the description of pathogenic variants background in Northern Brazil, as well as demonstrated the potential of a multigene panel in identifying pathogenic variants in genes not typically tested in hereditary cancer specific cases. The results obtained in this study observed 81 VUS, seven predicted with deleterious potential, reinforcing the need to identify pathogenicity of these variants in our population. These results had a great impact on the patients and their relatives since it allowed genetic counselling and personalized management decisions.
Availability of data and materials
The raw sequencing reads of all libraries were deposited at the European Nucleotide Archive [Accession number: PRJEB43823]. The datasets obtained from web-based sources and subsequently analysed in our study were: human genome (hg19) (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/), Mills & 1000G Gold Standard Indels (GATK resource bundle) (https://console.cloud.google.com/storage/browser/gcp-public-data%2D%2Dbroad-references/hg19/v0), dbSNP Build 138 (https://www.ncbi.nlm.nih.gov/snp/), dbNSFP database v3.5a (https://sites.google.com/site/jpopgen/dbNSFP) and ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/).
Abbreviations
- DGC:
-
Diffuse gastric cancer
- HBOC:
-
Hereditary breast and ovarian cancer
- HDGC:
-
Hereditary diffuse gastric cancer
- LBC:
-
Lobular breast cancer
- NGS:
-
Next generation sequencing
- SNVs:
-
Single nucleotides variants
- UTR:
-
Untranslated regions
- VUS:
-
Variant uncertain significance
References
Hereditary Cancer Syndromes and Risk Assessment. ACOG COMMITTEE OPINION, Number 793. Obstet Gynecol. 2019;134:e143–9.
Lu KH, Wood ME, Daniels M, Burke C, Ford J, Kauff ND, et al. American Society of Clinical Oncology expert statement: collection and use of a Cancer family history for oncology providers. J Clin Oncol. 2014;32(8):833–40. https://doi.org/10.1200/JCO.2013.50.9257.
Rich TA, Liu M, Etzel CJ, Bannon SA, Mork ME, Ready K, et al. Comparison of attitudes regarding preimplantation genetic diagnosis among patients with hereditary cancer syndromes. Familial Cancer. 2014;13(2):291–9. https://doi.org/10.1007/s10689-013-9685-0.
Rahner N, Steinke V. Hereditary Cancer syndromes. Dtsch Arztebl Int. 2008;105(41):706–14. https://doi.org/10.3238/arztebl.2008.0706.
Alemar B, Gregório C, Herzog J, Matzenbacher Bittar C, Brinckmann Oliveira Netto C, Artigalas O, et al. BRCA1 and BRCA2 mutational profile and prevalence in hereditary breast and ovarian cancer (HBOC) probands from Southern Brazil: Are international testing criteria appropriate for this specific population? PLoS One. 2017;12. https://doi.org/10.1371/journal.pone.0187630.
da Silva FCC, Valentin MD, de Ferreira FO, Carraro DM, Rossi BM. Mismatch repair genes in Lynch syndrome: a review. Sao Paulo Med J. 2009;127(1):46–51. https://doi.org/10.1590/s1516-31802009000100010.
Oliveira C, Senz J, Kaurah P, Pinheiro H, Sanges R, Haegert A, et al. Germline CDH1 deletions in hereditary diffuse gastric cancer families. Hum Mol Genet. 2009;18(9):1545–55. https://doi.org/10.1093/hmg/ddp046.
Sokolenko AP, Imyanitov EN. Molecular diagnostics in clinical oncology. Front Mol Biosci. 2018;5. https://doi.org/10.3389/fmolb.2018.00076.
Stanislaw C, Xue Y, Wilcox WR. Genetic evaluation and testing for hereditary forms of cancer in the era of next-generation sequencing. Cancer Biol Med. 2016;13(1):55–67. https://doi.org/10.20892/j.issn.2095-3941.2016.0002.
Okur V, Chung WK. The impact of hereditary cancer gene panels on clinical care and lessons learned. Cold Spring Harb Mol Case Stud. 2017;3(6):a002154. https://doi.org/10.1101/mcs.a002154.
Garber JE, Offit K. Hereditary Cancer predisposition syndromes. JCO. 2005;23(2):276–92. https://doi.org/10.1200/JCO.2005.10.042.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–60. https://doi.org/10.1093/bioinformatics/btp324.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4. https://doi.org/10.1093/bioinformatics/btv098.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1–11.10.33.
Mills RE, Luttig CT, Larkins CE, Beauchamp A, Tsui C, Pittard WS, et al. An initial map of insertion and deletion (INDEL) variation in the human genome. Genome Res. 2006;16(9):1182–90. https://doi.org/10.1101/gr.4565806.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. https://doi.org/10.1093/nar/29.1.308.
Liu X, Wu C, Li C, Boerwinkle E. dbNSFP v3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37(3):235–41. https://doi.org/10.1002/humu.22932.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92. https://doi.org/10.4161/fly.19695.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47(D1):D886–94. https://doi.org/10.1093/nar/gky1016.
Shihab HA, Gough J, Cooper DN, Stenson PD, Barker GLA, Edwards KJ, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden Markov models. Hum Mutat. 2013;34(1):57–65. https://doi.org/10.1002/humu.22225.
Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19(9):1553–61. https://doi.org/10.1101/gr.092619.109.
Kim S, Jhong J-H, Lee J, Koo J-Y. Meta-analytic support vector machine for integrating multiple omics data. BioData Min. 2017;10(1):2. https://doi.org/10.1186/s13040-017-0126-8.
Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011;39(17):e118. https://doi.org/10.1093/nar/gkr407.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2. https://doi.org/10.1038/nmeth.2890.
Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–7. https://doi.org/10.1093/bioinformatics/btv195.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9. https://doi.org/10.1038/nmeth0410-248.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31(13):3812–4. https://doi.org/10.1093/nar/gkg509.
Heald B, Marquard J, Funchain P. Strategies for clinical implementation of screening for hereditary cancer syndromes. Semin Oncol. 2016;43(5):609–14. https://doi.org/10.1053/j.seminoncol.2016.08.008.
Nyberg T, Frost D, Barrowdale D, Evans DG, Bancroft E, Adlard J, et al. Prostate Cancer risks for male BRCA1 and BRCA2 mutation carriers: a prospective cohort study. Eur Urol. 2020;77(1):24–35. https://doi.org/10.1016/j.eururo.2019.08.025.
Rizzolo P, Silvestri V, Tommasi S, Pinto R, Danza K, Falchetti M, et al. Male breast cancer: genetics, epigenetics, and ethical aspects. Ann Oncol. 2013;24:viii75–82.
Jarhelle E, Riise Stensland HMF, Hansen GÅM, Skarsfjord S, Jonsrud C, Ingebrigtsen M, et al. Identifying sequence variants contributing to hereditary breast and ovarian cancer in BRCA1 and BRCA2 negative breast and ovarian cancer patients. Sci Rep. 2019;9:1–12.
Kast K, Rhiem K, Wappenschmidt B, Hahnen E, Hauke J, Bluemcke B, et al. Prevalence of BRCA1/2 germline mutations in 21 401 families with breast and ovarian cancer. J Med Genet. 2016;53(7):465–71. https://doi.org/10.1136/jmedgenet-2015-103672.
Blair VR, McLeod M, Carneiro F, Coit DG, D’Addario JL, van Dieren JM, et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. The Lancet Oncology. 2020;21(8):e386–97. https://doi.org/10.1016/S1470-2045(20)30219-9.
Corso G, Intra M, Trentin C, Veronesi P, Galimberti V. CDH1 germline mutations and hereditary lobular breast cancer. Familial Cancer. 2016;15(2):215–9. https://doi.org/10.1007/s10689-016-9869-5.
Venesio T, Balsamo A, D’Agostino V, Ranzani GN. MUTYH-associated polyposis (MAP), the syndrome implicating base excision repair in inherited predisposition to colorectal tumors. Front Oncol. 2012;2. https://doi.org/10.3389/fonc.2012.00083.
Grover S, Kastrinos F, Steyerberg EW, Cook EF, Dewanwala A, Burbidge LA, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA. 2012;308(5):485–92. https://doi.org/10.1001/jama.2012.8780.
Rennert G, Lejbkowicz F, Cohen I, Pinchev M, Rennert HS, Barnett-Griness O. MutYH mutation carriers have increased breast cancer risk. Cancer. 2012;118(8):1989–93. https://doi.org/10.1002/cncr.26506.
Fulk K, LaDuca H, Black MH, Qian D, Tian Y, Yussuf A, et al. Monoallelic MUTYH carrier status is not associated with increased breast cancer risk in a multigene panel cohort. Familial Cancer. 2019;18(2):197–201. https://doi.org/10.1007/s10689-018-00114-4.
Win AK, Cleary SP, Dowty JG, Baron JA, Young JP, Buchanan DD, et al. Cancer risks for monoallelic MUTYH mutation carriers with a family history of colorectal cancer. Int J Cancer. 2011;129(9):2256–62. https://doi.org/10.1002/ijc.25870.
Hansford S, Kaurah P, Li-Chang H, Woo M, Senz J, Pinheiro H, et al. Hereditary diffuse gastric Cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015;1(1):23–32. https://doi.org/10.1001/jamaoncol.2014.168.
Oliveira C, Pinheiro H, Figueiredo J, Seruca R, Carneiro F. Familial gastric cancer: genetic susceptibility, pathology, and implications for management. Lancet Oncol. 2015;16(2):e60–70. https://doi.org/10.1016/S1470-2045(14)71016-2.
Capelle LG, Grieken NCTV, Lingsma HF, Steyerberg EW, Klokman WJ, Bruno MJ, et al. Risk and epidemiological time trends of gastric Cancer in lynch syndrome carriers in the Netherlands. Gastroenterology. 2010;138(2):487–92. https://doi.org/10.1053/j.gastro.2009.10.051.
Schneider NB, Pastor T, de Paula AE, Achatz MI, dos Santos ÂR, Vianna FSL, et al. Germline MLH1, MSH2 and MSH6 variants in Brazilian patients with colorectal cancer and clinical features suggestive of lynch syndrome. Cancer Medicine. 2018;7(5):2078–88. https://doi.org/10.1002/cam4.1316.
Dominguez-Valentin M, Nilbert M, Wernhoff P, López-Köstner F, Vaccaro C, Sarroca C, et al. Mutation spectrum in south American lynch syndrome families. Hereditary Cancer Clin Pract. 2013;11(1):18. https://doi.org/10.1186/1897-4287-11-18.
de Souza AM, Resende SS, de Sousa TN, de Brito CFA, de Souza AM, Resende SS, et al. A systematic sco** review of the genetic ancestry of the Brazilian population. Genet Mol Biol. 2019;42(3):495–508. https://doi.org/10.1590/1678-4685-gmb-2018-0076.
Caswell-** JL, Gupta T, Hall E, Petrovchich IM, Mills MA, Kingham KE, et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genetics in Medicine. 2018;20(2):234–9. https://doi.org/10.1038/gim.2017.96.
Kurian AW. BRCA1 and BRCA2 mutations across race and ethnicity: distribution and clinical implications. Curr Opin Obstet Gynecol. 2010;22(1):72–8. https://doi.org/10.1097/GCO.0b013e328332dca3.
Acknowledgements
We thank the patients that have agreed to participate of this study and the funding institutions, including Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES), Conselho Nacional de Desenvolvimento (CNPq) and Pró-Reitoria de Pesquisa e Pós-Graduação da Universidade Federal do Pará (PROPESP-UFPA). We also thank the Brazilian Society of Genetics (SBG) for presenting the abstract in their website.
Funding
This study was funded by Rede de Pesquisa em Genômica Populacional Humana (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - CAPES/Biologia Computacional: No. 3381/2013/CAPES) and Pró-Reitoria de Pesquisa e Pós-Graduação da Universidade Federal do Pará - PROPESP/UFPA. Â.R.S. was supported by Conselho Nacional de Desenvolvimento Científico e Tecnológico - CNPq/Productivity: 304413/2015–1. The funders had no role in the collection, analysis, interpretation of data or writing of the manuscript.
Author information
Authors and Affiliations
Contributions
AEH, AFV, ARS, CLBLP, WFB, PPA and SS contributed to the conception and design of the study. AFV, CSS, LM, MRM, RAV, and TVS performed the experiments. AMRS and LMB contributed to data collection and analysis of data. AFV and RSF wrote the manuscript. AEH and ARS performed the review of the final manuscript. All authors read and approved the manuscript.
Authors’ information
Amanda Ferreira Vidal and Rafaella Sousa Ferraz contributed equally to this work.
Affiliations.
Laboratory of Human and Medical Genetics, Graduate Program Genetics and Molecular Biology, Federal University of Pará, Belém, Pará, Brazil.
Amanda Ferreira Vidal, Rafaella Sousa Ferraz, Caio Santos Silva, Tatiana Vinasco-Sandoval, Leandro Magalhães, Milene Raiol-Moraes, Leonardo Miranda de Brito, Ricardo Assunção Vialle, Sidney Santos, Ândrea Ribeiro-dos-Santos & André M. Ribeiro-dos-Santos.
Bettina Ferro de Souza University Hospital, Federal University of Pará, Belém, Pará, Brazil.
Antonette El-Husny.
João de Barros Barreto University Hospital, Federal University of Pará, Belém, Pará, Brazil.
Williams Fernandes Barra & Cynthia Mara Brito Lins Pereira.
Center of Oncology Research, Federal University of Pará, Belém, Pará, Brazil.
Williams Fernandes Barra, Cynthia Lara Brito Lins Pereira, Paulo Pimentel de Assumpção, Sidney Santos & Ândrea Ribeiro-dos-Santos.
Corresponding author.
Correspondence to André M. Ribeiro-dos-Santos.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Review Board from Hospital Universitário João de Barros Barreto (HUJBB, Belém, Pará, Brazil) (CAAE: 89363618.3.0000.5634). All procedures performed in the present study involving human participants were conducted according to the ethical guidelines of Declaration of Helsinki. Written informed consent was obtained from all study participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1
Primers used for validation of pathogenic variants reported in the pan-cancer panel.
Additional file 2: Supplementary Table S2
List of variants of uncertain significance (VUS) reported in pan-cancer panel.
Additional file 3: Supplementary Table S3
Pathogenicity prediction of the variants of uncertain significance.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Vidal, A.F., Ferraz, R.S., El-Husny, A. et al. Comprehensive analysis of germline mutations in northern Brazil: a panel of 16 genes for hereditary cancer-predisposing syndrome investigation. BMC Cancer 21, 363 (2021). https://doi.org/10.1186/s12885-021-08089-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-021-08089-9