
Abstract
Background
Pruritus (itch) is a symptom commonly experienced by patients with cholestatic liver diseases such as primary biliary cholangitis (PBC, previously referred to as primary biliary cirrhosis). Bile acids (BAs) have been proposed as potential pruritogens in PBC. The ileal bile acid transporter (IBAT) protein expressed in the distal ileum plays a key role in the enterohepatic circulation of BAs. Pharmacological inhibition of IBAT with GSK2330672 may reduce BA levels in the systemic circulation and improve pruritus.
Methods
This clinical study (BAT117213 study) is sponsored by GlaxoSmithKline (GSK) with associated exploratory studies supported by the National Institute for Health Research (NIHR). It is a phase 2a, multi-centre, randomised, double bind, placebo controlled, cross-over trial for PBC patients with pruritus. The primary objective is to investigate the safety and tolerability of repeat doses of GSK2330672, and explore whether GSK2330672 administration for 14 days improves pruritus compared with placebo. The key outcomes include improvement in pruritus scores evaluated on a numerical rating scale and other PBC symptoms in an electronic diary completed twice daily by the patients. The secondary outcomes include the evaluation of the effect of GSK2330672 on total serum bile acid (BA) concentrations, serum markers of BA synthesis and steady-state pharmacokinetics of ursodeoxycholic acid (UDCA).
Discussion
BAT117213 study is the first randomised controlled crossover trial of ileal bile acid transporter inhibitor, a novel class of drug to treat pruritus in PBC. The main strengths of the trial are utility of a novel, study specific, electronic symptom diary as patient reported outcome to measure the treatment response objectively and the crossover design that allows estimating the treatment effect in a smaller number of patients. The outcome of this trial will inform the trial design of future development phase of the IBAT inhibitor drug. The trial will also provide opportunity to conduct metabonomic and gut microbiome studies as explorative and mechanistic research in patients with cholestatic pruritus.
Trial registration
EudraCT number: 2012-005531-84, ClinicalTrials.gov Identifier: NCT01899703, registered on 3rd July 2013
Similar content being viewed by others


Background
Primary biliary cholangitis (cirrhosis) (PBC) is an autoimmune chronic cholestatic liver disease with a prevalence of 30/100,000, typically affecting middle aged women (female: male ratio 10:1) [1]. In untreated cases immunologically mediated chronic cholestasis ultimately results in liver cirrhosis with associated complications such as portal hypertension, varices, ascites, hepatocellular carcinoma and death. The precise aetiology of PBC is unclear, although genetic and environmental factors are thought to play a key role.
Pruritus (itch) is one of the characteristic symptoms of PBC and can affect patients at any stage of the disease [2]. Recently, we studied the scale of the pruritus symptom within the United Kingdom (UK)-PBC cohort, a national cohort of over 3000 PBC patients recruited from every hospital in the UK. In this cohort 60–70 % of PBC patients reported experience of pruritus at some point in the course of the disease, 30 % had persistent pruritus and 15 % suffered with severe pruritus since the diagnosis of PBC [3]. A similar scale of symptom burden has also been reported in PBC cohorts from USA and Italy [4]. Pruritus has a negative impact on perceived quality of life in PBC patients and has been associated with sleep deprivation, worsened day time fatigue and when severe, may lead to depression and suicidal tendencies [5].
Ursodeoxycholic acid (UDCA), the current standard of care for PBC patients and the only licenced therapy for PBC has no role in treating pruritus [2]. Current treatment of pruritus in PBC involves step-wise use of specific anti-pruritic agents in line with current international guidelines [2, 6]. These drugs include cholestyramine, rifampicin, naltrexone and sertraline. Of these, cholestyramine is the only licensed drug for treatment of cholestatic pruritus and use of other drugs is “off-label”. The limitations of these drugs are that their efficacy is not universal, treatment is often associated with side effects and there is a need for regular monitoring for liver toxicity. Patients with medically refractory pruritus may either need to undergo phototherapy, invasive interventions such as nasobiliary drainage or extracorporeal albumin dialysis for temporary relief of pruritus, or may be considered for liver transplantation (LT) which is typically curative. Therefore, development of better drug therapies with fewer side effects is an unmet clinical need for PBC patients [7].
Ileal bile acid transporter (IBAT)
Primary BAs are synthesized in the liver from an enzymatic catabolism of cholesterol, a process regulated by enzyme cytochrome P450 (CYP) 7A1. Unconjugated BAs are conjugated in hepatocytes with glycine and taurine, secreted into the bile and stored in the gallbladder. Upon ingestion of a meal, conjugated BAs (“bile salts”) are released into the intestinal lumen where they facilitate absorption of fat and fat soluble vitamins. After their normal physiological function is completed in the intestine, BAs reach the ileum where they are reabsorbed. The ileal bile acid transporter [(IBAT), also called apical sodium dependent bile acid transporter (ASBT)], is a protein predominantly located in the terminal ileum and serves as the main transporter mediating the ileal uptake of conjugated BAs and their return to the liver via the portal circulation (enterohepatic circulation) [8].
Bile salts (and their protonated form, BAs) have been suggested to play role in the pathogenesis of pruritus in cholestatic conditions. In cholestasis, the ileal uptake of BAs has been shown to be upregulated [9]. Also, the evidence that pruritus dramatically improves in patients undergoing nasobiliary drainage [10] and is effectively cured by LT [11] suggests a direct or indirect role for BAs in mediating cholestatic pruritus. Therefore a pharmaceutical agent that can reduce their levels in the enterohepatic and systemic circulations may be predicted to improve pruritus. In two animal studies treatment with IBAT inhibitors SC-435 and A4250 produced BA malabsorption and attenuated BA-mediated cholestatic liver injury by reducing biliary BA output [12, 13]. In humans, use of IBAT inhibitor A4250 has been shown to decrease the serum BAs and increase faecal BAs by highly efficient interruption of their enterohepatic circulation with no serious adverse events [14].
GSK2330672
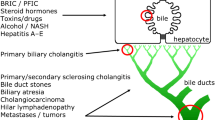
GSK2330672 is a selective inhibitor of human IBAT and it is designed to be a non-absorbable agent restricted to the gastrointestinal (GI) tract. GSK2330672 is expected to block the uptake of BAs in the terminal ileum, increase their excretion in the faeces and decrease the amount of BAs returning to the liver via enterohepatic circulation (Fig. 1). Therefore treatment of PBC patients with oral GSK2330672 is postulated to reduce concentrations of BAs in the systemic circulation and in turn improve pruritus.

Mechanism of IBAT (ASBT) inhibitor drug. GSK2330672 interrupts the enterohepatic circulation of bile acids by selectively inhibiting the IBAT protein located in the terminal ileum, thereby reducing the levels of bile acids in the systemic circulation. (Image reproduced with permission from [31]). IBAT, ileal bile acid transporter; ASBT, apical sodium-dependent bile acid transporter
In phase I studies involving 42 healthy volunteers single and repeat doses of GSK2330672 for 12 days were shown to be safe and tolerable (ClinicalTrials.gov Identifier: NCT01416324). GI symptoms were the most common reported drug-related adverse events (AEs). These included diarrhoea, abdominal pain, bowel movement irregularity and positive faecal occult blood tests. All AEs were considered mild or moderate in severity.
Methods
Study design and overview
The BAT117213 study is a Phase 2a trial, designed to investigate treatment with GSK2330672 in PBC patients with pruritus (ClinicalTrials.gov Identifier: NCT01899703). This is a multicentre, randomized, double-blind, placebo-controlled, two-period cross-over trial which in addition to studying the safety and efficacy of the drug will provide an opportunity to conduct explorative studies (including metabonomic and microbiomic studies) to develop novel mechanistic insights into cholestatic pruritus.
Following written informed consent, patients with PBC and pruritus were screened to establish study eligibility. Eligible subjects participated in a two-week placebo run-in period followed by randomization in a crossover fashion to receive placebo or GSK2330672 treatment during two consecutive two-week study periods (Sequence 1/Sequence 2) (Fig. 2). Subjects then participated in a two-week follow up period of placebo dosing. Total duration of the study was 56 days from the first day of dosing.

Flowchart of BAT117213 study design
Study population
The study population consisted of PBC patients with ongoing pruritus. All participants had a diagnosis of definite or probable PBC established according to recognised criteria [2, 6]. Key inclusion and exclusion criteria for study eligibility are detailed in Table 1. The trial entry criteria for ongoing pruritus was defined as: i) severe pruritus significantly impacting daily life and proven refractory to medical therapy, or ii) pruritus that is newly diagnosed or untreated, or iii) pruritus that is unresolved with the use of a single antipruritic agent. To determine subject eligibility for study enrolment outpatient screening was performed within 45 days before the first dose administration. Subjects meeting all the inclusion criteria and no exclusion criteria were enrolled by a designated investigator from the centre.
Study objectives and outcomes
The primary objective of this trial is to investigate the safety and tolerability of oral GSK2330672 compared with placebo when administered for 14 days to PBC patients treated with UDCA. The secondary objectives are: 1) to evaluate the effects of oral GSK2330672 on subjects’ experience of pruritus and its impact; 2) to demonstrate the lack of effect of oral GSK2330672 on steady-state pharmacokinetics (PK) of UDCA when UDCA is administered alone or in combination with GSK2330672; 3) to investigate the steady state PK of oral GSK2330672; 4) to evaluate the effects of oral GSK2330672 on total serum BA concentrations and serum markers of BA synthesis [7-alpha-hydroxy-4-cholesten-3-one (C4)]. Exploratory objectives of the study include investigating effects of 14-day oral administration of GSK2330672 on markers of disease progression, subject’s experience of benefits and disadvantages with GSK2330672, metabonomics, microbiomics and pharmacogenomics. The primary, secondary and exploratory outcome measures are given in Table 2.
Recruitment and consent
The study is a UK multicentre study and recruitment was planned in three large, tertiary referral National Health Service (NHS) hospitals based in Newcastle, Birmingham and Cambridge. Patients were recruited from the out-patient department cohorts of these hospitals and in addition, trial information was published in newsletters and magazines from the UK-PBC research group and patient support groups (LIVErNORTH and PBC Foundation). Any PBC patient interested in participating in the study could contact the study team at the centre nearest to their location either directly or via referral from local primary or secondary care physicians. The UK-PBC platform was utilised for recruitment using a similar approach to the to the RIT-PBC trial reported recently by our group [15]. The established UK-PBC database was screened for patients with PBC-40 itch domain scores meeting the definitions of persistent and/or severe pruritus. The clinicians looking after these patients were contacted to approach the patients and interested patients were referred to their local recruiting centre. All participants gave their written consent to participation before screening investigations were performed. Participants completed the consent process with study investigators trained in Good Clinical Practice (GCP) and assessment of capacity.
Randomisation
All eligible subjects enrolled in the study were randomised to either Sequence 1 or Sequence 2 to receive oral placebo or GSK2330672 for a 14-day period in a cross-over fashion (Fig. 2). Sequence 1 was GSK2330672 for 14 days followed by placebo for 14 days and Sequence 2 was placebo for 14 days followed by GSK2330672 for 14 days. Randomisation was carried out via a dedicated electronic system for randomisation-RAMOS (Randomisation and Medication Ordering System) by generating a unique randomisation number for each participant that linked to the corresponding allocated sequence of study drug.
Study treatment
The investigational medicinal product used in this study was GSK2330672. The control intervention was placebo. Both GSK233072 and placebo were manufactured at a dedicated manufacturing unit in London (UK) and dispensed as 30 g aliquots of oral solution into amber glass bottles for distribution to participating study centres. The study centres supplied solutions to subjects in accordance with the randomization schedule. Subjects consumed the entire quantity of one or two bottles of study drug twice daily followed by two 50 mL rinses of water. All patients started the study with 14-days placebo run in period followed by 14-days treatment with GSK2330672 or placebo in a cross over fashion.
Dose escalation and stop** criteria
The initial dose of GSK2330672 was 45 mg and all patients were asked to increase the dose to 90 mg on day 4. If this was not tolerated, they were asked to continue at 45 mg and attempt a dose increase again two days later. If 90 mg could not be tolerated by the end of day 7, subjects were asked to continue only 45 mg.
Following stop** criteria were in place to assure subject safety: 1) to stop the study treatment if the stop** criteria for liver chemistry were met [ALT >5-8 x upper limit of normal (ULN), bilirubin > 1.5-2 x ULN], and 2) to withdraw the subject from the study if corrected QT (QTc) interval withdrawal criteria were met based on their average values on triplicate ECGs separated by five minutes. These were QTc > 500 msec, or uncorrected QT >600 msec, or QTc >60 msec change from baseline). If a subject met the stop** criteria, appropriate safety follow-up assessments and procedures were completed.
Concomitant medications
Before starting the study, all patients were advised to stop using their usual anti-pruritic agents including cholestyramine, colesevelam, rifampicin, naltrexone, sertraline, gabapentin and anti-histamines. The use of these medications was prohibited during the study period until the final follow-up period when rescue medications were permitted. Application of topical agents used to relieve pruritus was permitted during the study only if agents did not contain active ingredients in the list of prohibited agents and with prior agreement of the clinical investigator. Subjects were asked to abstain from taking new prescription or new non-prescription drugs (including vitamins and dietary or herbal supplements), from the start of the placebo run-in period until completion of the follow-up visit. The use of UDCA was permitted and patients who were on UDCA were standardised to receive Ursofalk®(Dr. Falk Pharma UK Ltd) once daily preparation at dose 13–15 mg/kg/day and instructed to take it at bed time.
Patient reported outcomes
Existing patient reported outcome (PRO) measures to assess the impact of PBC symptoms include the PBC-40, a widely acceptable, validated, disease-specific questionnaire and the 5-D Itch scale [16, 17]. However, for this study a more specific PRO measure was needed that could detect the severity and variability of pruritus and other PBC symptoms and potential treatment effects on a daily basis with a short recall period. The development of such a measure began with interviews with PBC patients to identify additional characteristics of pruritus and other symptoms and their impact on sleep and daily activities. With input from PBC patients and PRO experts a new electronic patient reported outcome (ePRO) diary was developed to assess the severity of the pruritus and other PBC symptoms. Subjects completed the ePRO diary every morning and evening before dosing the study drug. In the ePRO diary pruritus severity was rated using a numerical rating scale (NRS). Psychometric testing to support the validity and reliability of the ePRO will be evaluated with data from the current clinical study.
Data analysis
Statistical analysis
This trial is designed to estimate the effect of study drug GSK2330672 relative to placebo when co-administered with UDCA on pruritus symptom, markers of efficacy and disease progression and the PK of UDCA. No formal hypothesis will be tested.
The efficacy endpoint in this study is the patient reported rating of pruritus severity scores. Pruritus will be measured in three different PROs: pruritus NRS using the ePRO, the 5-D itch scale and the PBC-40 questionnaire [16, 17]. Changes in pruritus NRS will be used as the key measure of the efficacy endpoint and will be analysed using a mixed effects model with fixed effect terms for treatment period and sequence to examine differences between GSK2330672 and placebo. Subject will be treated as a random effect in the model. Point estimates and their associated 95 % confidence interval (CI) will be constructed for the mean differences in pruritus severity scores.
Data from subjects that are co-administered UDCA as part of their standard care will be analysed similarly for PK endpoints. Following log-transformation, maximum observed plasma concentration (Cmax), AUC (0–12 h) and AUC (12–24 h) of UDCA and glycine and taurine conjugated metabolites of UDCA (TUDCA and GUDCA) will be separately analysed. This will be done using a mixed effects model with fixed effect terms for treatment period and sequence to examine differences between UDCA administration with and without GSK2330672. Point estimates and corresponding 90 % CI will also be constructed for the difference and/or ratio between the mean of the test treatment (UDCA plus GSK2330672) and the mean of the reference treatment (UDCA alone).
Sample size
The efficacy endpoint in this study is pruritus score and the sample size for efficacy endpoint is based on the pruritus 0 to 10 points scale. On this scale the average effect of rifampicin is 1.62 points and the reported pooled total standard deviations of various anti-pruritic drugs ranges from 1.22 to 3.84 points [18, 19]. Assuming that GSK2330672 is at least as effective as rifampicin, a sample size of 40 will result in a reasonable power (>90 %) if the standard deviation (SD) is 3.1 points or less. For estimation of relative bioavailability 20 subjects taking UDCA are required to ensure that the resultant 90 % CI of the ratio will be within 0.8 and 1.25 assuming that the true ratio is 1 and the SD on the log10 scale is less than 0.25.
An initial sample-size of 40 subjects was selected based on considerations of both efficacy and PK endpoints. However, due to the uncertainty around sample-size assumptions a series of interim analyses for futility and possible sample-size re-estimation were carried out at regular intervals. Data from completed patients were reviewed by an unblinded review committee (composed of GSK personnel not directly involved in study conduct). As the probability of demonstrating sufficient difference was high, the sponsor revised the sample-size from 40 to 22. No other changes to study conduct were planned as a result of the interim analyses.
Conduct of the trial
The conduct of the trial followed the principles outlined in the NHS research governance framework for health and social care, GCP and the guiding principles of the 2008 Declaration of Helsinki. The trial involved the participant visiting the study centre a total of six times including screening visit, day 1 visit, three consecutive fortnightly in-patient stays (each up to 36 h) and a follow up visit. The schedule of study procedures during these visits and data collection is summarised in Table 3.
Protocol deviation or exemptions were not allowed with the exception of immediate safety concerns. All Investigators at recruiting sites followed standard operative procedures for collection, handling, processing and storage of samples (blood, urine and stool) collected at study visits. All clinical and non-clinical subject data including medical history (to capture co-morbidities and concomitant medications) and physical examinations were entered into electronic case report forms (eCRFs). No patient identifiable information was entered in the eCRFs. All participants were allocated a unique study identifier which was used on eCRFs transmitted electronically to the sponsor and combined with data provided from other sources in a validated data system.
Study monitoring
The study sponsor performed periodic monitoring at each study centre to monitor the study conduct and site activity. The monitor had direct access to all relevant documents to verify the data for completeness, accuracy and authenticity and the site’s compliance with study protocol. All monitoring findings were reported and followed up in a timely manner. Periodic interim analysis of the trial were undertaken to determine as to whether the study should be modified, continued or terminated.
Adverse events
AEs and serious adverse events (SAEs) were collected from the start of the placebo run-in period (day 1) until the follow-up contact (day 56). The investigator and site staff were responsible for detecting, documenting and reporting events that meet the definition of an AE or SAE. All SAEs were recorded and reported to the study sponsor within 24 h. Periodic reviews of the safety data were performed and presented during interim analysis to both the sponsor and the study investigators.
Sponsorship, insurance and indemnity
In accordance with the Association of the British Pharmaceutical Industry (ABPI) guidance, the trial sponsor had policies in place regarding compensation for any trial related harm due to negligence or otherwise. The trial sponsor had insurance to cover indemnity in respect of potential liability arising from negligent harm related to study design. Due to the commercial nature of the study there were also arrangements for non-negligent compensation. The participating study centres were NHS hospitals and the NHS indemnity covered NHS staff and medical academic staff with honorary NHS contracts conducting the study for potential liability in respect of negligent harm arising from the conduct of the study.
Trial status
The BAT117213 study was opened for recruitment in January 2014 with first patient recruited in March 2014. The initial recruitment target was 40 subjects. Following review of safety and efficacy of data from 11 patients at the first interim analysis in March 2015, the sponsor decided to continue the study recruitment. A second interim analysis of the data from 19 patients was performed in July 2015 and the sponsor decided to reduce the total sample size to 22 patients. The recruitment ended in October 2015 with all 22 patients randomised from two trial sites (Newcastle 13; Birmingham 9). The treatment follow-up of participants was completed in December 2015. The analysis of study data is currently ongoing and results are scheduled to be available in November 2016.
Discussion
Need for novel anti-pruritic drugs in PBC
Pruritus is a complex symptom and the drug treatment of pruritus in PBC patients remains a challenge in clinical practice. The four main classes of drugs that are recommended by current guidelines [2, 6] include bile acid sequestrants (cholestyramine), enzyme inducers (rifampicin), opioid antagonists (naltrexone) and selective serotonin re-uptake inhibitors (sertraline). These drugs are limited by their lack of universal efficacy, poor compliance (especially cholestyramine) and the need for regular monitoring for liver toxicity (rifampicin). Cholestyramine and rifampicin have good reports but clinical experience of both naltrexone and sertraline has been disappointing for many clinicians [2].
A critical review of literature shows that the strength of evidence for current anti-pruritic drug therapy is poor. Cholestyramine, the current first-line therapy was last studied over five decades ago but has never been subjected to randomised placebo-controlled trials and has evidence category II-2 (cohort or case control analytical studies) [20–24]. Only rifampicin and naltrexone have been studied in controlled trials [18, 19, 25–27] and sertraline (evidence category II-2) is the last agent investigated with a positive outcome on pruritus [28]. A number of other drugs have been investigated but with little success and more recently both gabapentin (2006) and colesevelam (2010) trials failed to show any therapeutic benefit in cholestatic pruritus [29, 30].
IBAT2330672 trial
The apparent lack of novel drug development in cholestatic pruritus can be attributed partly to incomplete understanding of the complex pathophysiology of the disease. More recent advances in molecular research have identified novel targets for drug development in cholestasis. IBAT inhibitors are novel class of drugs with therapeutic potential in cholestasis. They have been shown be beneficial in cholestasis by the experimental studies and their desired effects on serum and faecal bile acid profile has been proven in healthy people [13, 14].
The BAT117213 study is the first phase 2 multicentre, double-blinded, placebo-controlled crossover trial designed to investigate the safety and efficacy of IBAT inhibitor in PBC patients with pruritus. Unlike the only other phase 2 trial of an IBAT inhibitor drug (LUM001) in PBC (CLARITY study, NCT01904058), the main strength of the BAT117213 study is its crossover design which allows estimating the treatment effect in a smaller number of patients and reduces the between-patient variability and yields a more efficient comparison of treatments than a similar sized parallel group trial. In the BAT117213 study every patient will receive both the study drug and the placebo; therefore each patient will serve as his/her own matched control.
An additional strength of this trial is the utility of patient reported outcomes to measure the treatment response objectively using existing validated tools including the PBC-40 questionnaire and 5-D itch scale as well as a novel, easy-to-use electronic symptom diary. The latter has been specifically developed for this study and it contains morning and evening diaries with questions on itch, fatigue and concentration to comprehensively capture the severity of the symptoms over the preceding 12 h. In addition, the exit interviews conducted at the end of the study provide the opportunity for patients to express their experiences in the study in a semi-structured method that may not have been detected with the more structured patient reported outcomes measures.
The BAT117213 study also provides a unique opportunity to conduct novel, explorative, mechanistic research in patients with cholestatic pruritus. Serum and urine samples obtained during the study will be used to study the metabolic phenotype (metabonomics) of pruritus in PBC by using 1H (proton)-nuclear magnetic resonance (NMR) spectroscopy and mass spectrometry (MS). Similarly, using the faecal samples from study patients gut-microbiome studies will be undertaken to study the association between gut microbiota composition and pruritus in PBC. Results of these metabonomic and microbiomic studies are likely to provide more insight into the biology of pruritus in PBC and may identify potential biomarkers for cholestatic pruritus.
The main drawback of this trial is the potential carryover effect (i.e. effect of the treatment from the previous time period may “carry over” on the response to subsequent period) and lack of “washout period” between treatment periods. Carryover effect is a common problem inherent to the cross over study design and may potentially confound direct estimates of treatment effect. Therefore the statistical analysis the data will be assessed for any evidence of carry over and appropriate sensitivity analyses will be performed. To mitigate against the lack of “washout period” the outcome measurements will be restricted to the latter part of each treatment period.
In summary, BAT117213 study is a phase 2 study to evaluate the safety and tolerability of a unique class of drug in treating pruritus in PBC patients and provide novel information about bile acids and metabolic changes and gut microbiome profile in cholestatic pruritus. The results from this trial will inform the trial design of future development phase of the IBAT inhibitor drug.
Abbreviations
ABPI, association of the British Pharmaceutical Industry; AE, adverse event; ALT: alanine transaminase; ASBT, apical sodium dependent bile acid transporter; AUC, area under the curve; BA, bile acid; C4, 7-alpha-hydroxy-4-cholesten-3-one; CI, confidence interval; CYP, cytochrome P450; ECG, electrocardiogram; eCRF, electronic case report form; ePRO, electronic patient reported outcome; GCP, good clinical practice; GI, gastrointestinal; GSK, GlaxoSmithKline; GUDCA, glyco ursodeoxycholic acid; IBAT, Ileal bile acid transporter; LT, liver transplantation; MS, mass spectrometry; NHS, National Health Service (of the UK); NMR, nuclear magnetic resonance; NRS, numerical rating scale; PBC, primary biliary cholangitis (previously cirrhosis); PGX, pharmacogenetics; PK, pharmacokinetics; PRO, patient reported outcome; RAMOS, randomisation and medication ordering system; RCT, randomised controlled trial; SAE, serious adverse event; SD, standard deviation; TUDCA, tauro ursodeoxycholic acid; UDCA, ursodeoxycholic acid; ULN, upper limit of normal.
References
Griffiths L, Dyson JK, Jones DE. The new epidemiology of primary biliary cirrhosis. Semin Liver Dis. 2014;34(3):318–28. doi:10.1055/s-0034-1383730. published Online First: Epub Date.
Beuers U, Boberg KM, Chapman RW, et al. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237–67. doi:10.1016/j.jhep.2009.04.009. published Online First: Epub Date.
Hegade VS, Mells GF, Beuers U, et al. Patient Experience and Characteristics of Cholestatic Pruritus in the UK-PBC Research Cohort. Hepatology. 2014;60:339A–69. doi:10.1002/hep.27496. published Online First: Epub Date.
Hegade VS, Mells GF, Lammert C, et al. A Comparative Study of Pruritus in PBC cohorts from UK, USA and Italy. J Hepatol. 2015;62:S785.
Mells GF, Pells G, Newton JL, et al. Impact of primary biliary cirrhosis on perceived quality of life: the UK-PBC national study. Hepatology. 2013;58(1):273–83. doi:10.1002/hep.26365. published Online First: Epub Date.
Lindor KD, Gershwin ME, Poupon R, et al. Primary biliary cirrhosis. Hepatology. 2009;50(1):291–308. doi:10.1002/hep.22906. published Online First: Epub Date.
Dyson JK, Webb G, Hirschfield GM, et al. Unmet clinical need in autoimmune liver diseases. J Hepatol. 2015;62(1):208–18. doi:10.1016/j.jhep.2014.09.010. published Online First: Epub Date.
Dawson PA, Haywood J, Craddock AL, et al. Targeted deletion of the ileal bile acid transporter eliminates enterohepatic cycling of bile acids in mice. J Biol Chem. 2003;278(36):33920–7. doi:10.1074/jbc.M306370200. published Online First: Epub Date.
Lanzini A, De Tavonatti MG, Panarotto B, et al. Intestinal absorption of the bile acid analogue 75Se-homocholic acid-taurine is increased in primary biliary cirrhosis, and reverts to normal during ursodeoxycholic acid administration. Gut. 2003;52(9):1371–5.
Hegade VS, Krawczyk M, Kremer AE, et al. The safety and efficacy of nasobiliary drainage in the treatment of refractory cholestatic pruritus: a multicentre European study. Aliment Pharmacol Ther. 2016;43(2):294–302. doi:10.1111/apt.13449. published Online First: Epub Date.
Neuberger J, Jones EA. Liver transplantation for intractable pruritus is contraindicated before an adequate trial of opiate antagonist therapy. Eur J Gastroenterol Hepatol. 2001;13(11):1393–4.
Wong MH, Oelkers P, Dawson PA. Identification of a mutation in the ileal sodium-dependent bile acid transporter gene that abolishes transport activity. J Biol Chem. 1995;270(45):27228–34.
Baghdasaryan A, Fuchs CD, Osterreicher CH, et al. Inhibition of intestinal bile acid absorption improves cholestatic liver and bile duct injury in a mouse model of sclerosing cholangitis. J Hepatol. 2016;64(3):674–81. doi:10.1016/j.jhep.2015.10.024. published Online First: Epub Date.
Graffner H, Gillberg PG, Rikner L, Marschall HU. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment Pharmacol Ther. 2016;43(2):303–10. doi:10.1111/apt.13457. published Online First: Epub Date.
Jopson L, Newton JL, Palmer J, et al. RITPBC: B-cell depleting therapy (rituximab) as a treatment for fatigue in primary biliary cirrhosis: study protocol for a randomised controlled trial. BMJ Open. 2015;5(8):e007985. doi:10.1136/bmjopen-2015-007985. published Online First: Epub Date.
Jacoby A, Rannard A, Buck D, et al. Development, validation, and evaluation of the PBC-40, a disease specific health related quality of life measure for primary biliary cirrhosis. Gut. 2005;54(11):1622–9. doi:10.1136/gut.2005.065862. published Online First: Epub Date.
Elman S, Hynan LS, Gabriel V, Mayo MJ. The 5-D itch scale: a new measure of pruritus. Br J Dermatol. 2010;162(3):587–93. doi:10.1111/j.1365-2133.2009.09586.x. published Online First: Epub Date.
Khurana S, Singh P. Rifampin is safe for treatment of pruritus due to chronic cholestasis: a meta-analysis of prospective randomized-controlled trials. Liver Int. 2006;26(8):943–8. doi:10.1111/j.1478-3231.2006.01326.x. published Online First: Epub Date.
Tandon P, Rowe BH, Vandermeer B, Bain VG. The efficacy and safety of bile Acid binding agents, opioid antagonists, or rifampin in the treatment of cholestasis-associated pruritus. Am J Gastroenterol. 2007;102(7):1528–36. doi:10.1111/j.1572-0241.2007.01200.x. published Online First: Epub Date.
Datta DV, Sherlock S. Cholestyramine for long term relief of the pruritus complicating intrahepatic cholestasis. Gastroenterology. 1966;50(3):323–32.
Oster ZH, Rachmilewitz EA, Moran E, Stein Y. Relief of pruritus by cholestyramine in chronic liver disease. Isr J Med Sci. 1965;1(4):599–606.
Datta DV, Sherlock S. Treatment of pruritus of obstructive jaundice with cholestyramine. Br Med J. 1963;1(5325):216–9.
Van Itallie TB, Hashim SA, Crampton RS, Tennent DM. The treatment of pruritus and hypercholesteremia of primary biliary cirrhosis with cholestyramine. N Engl J Med. 1961;265:469–74. doi:10.1056/NEJM196109072651004. published Online First: Epub Date.
Carey Jr JB, Williams G. Relief of the pruritus of jaundice with a bile-acid sequestering resin. JAMA. 1961;176:432–5.
Terg R, Coronel E, Sorda J, Munoz AE, Findor J. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37(6):717–22.
Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Van Buuren HR. Oral naltrexone treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology. 1997;113(4):1264–9.
Ghent CN, Carruthers SG. Treatment of pruritus in primary biliary cirrhosis with rifampin. Results of a double-blind, crossover, randomized trial. Gastroenterology. 1988;94(2):488–93.
Mayo MJ, Handem I, Saldana S, Jacobe H, Getachew Y, Rush AJ. Sertraline as a first-line treatment for cholestatic pruritus. Hepatology. 2007;45(3):666–74. doi:10.1002/hep.21553. published Online First: Epub Date.
Bergasa NV, McGee M, Ginsburg IH, Engler D. Gabapentin in patients with the pruritus of cholestasis: a double-blind, randomized, placebo-controlled trial. Hepatology. 2006;44(5):1317–23. doi:10.1002/hep.21370. published Online First: Epub Date.
Kuiper EM, van Erpecum KJ, Beuers U, et al. The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: results of a double-blind, randomized, placebo-controlled trial. Hepatology. 2010;52(4):1334–40. doi:10.1002/hep.23821. published Online First: Epub Date.
Hegade VS, Kendrick SF, Jones DE. Drug treatment of pruritus in liver diseases. Clin Med. 2015;15(4):351–7. doi:10.7861/clinmedicine.15-4-351. published Online First: Epub Date.
Acknowledgments
We thank all the subjects for participating in this clinical trial and the NIHR Newcastle Biomedical Research Centre for supporting the exploratory studies associated with this clinical trial. VSH would like to thank NIHR Newcastle BRC for funding his clinical PhD fellowship in cholestatic pruritus. MC and GMH acknowledge support from NIHR Birmingham Liver Biomedical Research Unit. All authors would like to thank LIVErNORTH (www.livernorth.org.uk) and PBC Foundation (www.pbcfoundation.org.uk) for their ongoing support of trials in PBC and assistance in raising awareness of the trial and recruiting patients.
Funding
This clinical trial was funded by GlaxoSmithKline (GSK) [study number 117413]. The explorative studies associated with the clinical trial were funded by the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre [grant number BH134330].
Availability of data and materials
Not applicable.
Authors’ contributions
SFWK, RLD, DR and GD designed the study and developed the trial protocol. DEJJ, GMH and GA are principal investigators (PIs) of this study and advised on the study design and protocol. JS was involved in trial coordination. SRM as the trial statistician did the sample size calculation and analysis plan. KG and SV designed and developed the electronic diary. VSH and MC managed the trial. VSH was the sub-investigator of the participating centre and wrote the manuscript for this publication. All authors read and approved the final manuscript.
Competing interests
SFWK, RLD, SRM, DR, JS, GD, KG and SV are GlaxoSmithKline (GSK) employees. GMH and DEJJ are investigators on the UK-PBC consortium which has received research funding from GSK. VSH and MC have no competing interest to declare.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The National Research Ethics Service (NRES) Committee North East and Sunderland (REC reference 13/NE/0290) and the Medicine and Healthcare products Regulatory Agency (MHRA) approved all versions of the study protocol. In addition, all recruitment sites obtained approval from their respective hospital Research and Development (R&D) departments before starting screening patients. This study was conducted according to the ethical principles that have their origin in the Declaration of Helsinki and the Good Clinical Practice Guidelines (ICH-GCP).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hegade, V.S., Kendrick, S.F.W., Dobbins, R.L. et al. BAT117213: Ileal bile acid transporter (IBAT) inhibition as a treatment for pruritus in primary biliary cirrhosis: study protocol for a randomised controlled trial. BMC Gastroenterol 16, 71 (2016). https://doi.org/10.1186/s12876-016-0481-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12876-016-0481-9