
Abstract
Background
As an important nosocomial pathogen, Enterococcus faecium has received increasing attention in recent years. However, a large number of studies have focused on the hospital-associated isolates and ignored isolates originated from the natural environments.
Results
In this study, comparative genomic analysis was conducted on 161 isolates originated from human, animal, and naturally fermented dairy products. The results showed that the environment played an important role in sha** the genomes of Enterococcus faecium. The isolates from human had the largest average genome size, while the isolates from dairy products had the smallest average genome size and fewest antibiotic resistance genes. A phylogenetic tree was reconstructed based on the genomes of these isolates, which revealed new insights into the phylogenetic relationships among the dairy isolates and those from hospitals, communities, and animals. Furthermore, 202 environment-specific genes were identified, including 136 dairy-specific, 31 human blood-specific, and 35 human gastrointestinal-specific genes. Interestingly, five dairy-specific genes (namely lacF, lacA/B, lacD, lacG, and lacC) that constituted an integrated lactose metabolism pathway existed in almost all dairy isolates. The pathway conservation demonstrated an active role of the environment in sha** the genomes of Enterococcus faecium.
Conclusions
This study shows that the Enterococcus faecium species has great genomic plasticity and high versatility to occupy broad ecological roles, dwelling as non-harmful dairy and animal gut commensals as well as significant nosocomial pathogens that disseminate antibiotic resistance genes.
Similar content being viewed by others

Background
Enterococci are common gastrointestinal (GI) commensal bacteria in humans and other animals [1]. However, over the last 30 years, nosocomial infections due to enterococci have been increasing continuously and have become a top leading cause of hospital-acquired infections of the GI tract, bloodstream, and urinary tract (UT) [2,3,4,5]. Two Enterococcus (E.) species, E. faecalis and E. faecium, are of major clinical concerns. In the late 1970s and 1980s, hospital-acquired enterococcal infections were mainly caused by E. faecalis due to their high intrinsic virulence [2]. However, the prevalence of nosocomial infections caused by E. faecium is rising since the early 1990s in the hospitals in the United States, and it has even partially replaced E. faecalis to become a main culprit of nosocomial infections around the world in recent years [2, 6]. The major reason for E. faecium to become a top nosocomial pathogen is its multidrug resistance. Many of the modern nosocomial E. faecium isolates are resistant to ampicillin, aminoglycosides, quinupristin–dalfopristin, linezolid, and vancomycin [2]. Vancomycin is the last line of defense against a wide range of multi-resistant Gram-positive pathogens [7]. In recent years, the number of infections associated with vancomycin-resistant enterococci has increased rapidly in many countries. For example, the percentage of vancomycin-resistant E. faecium rose from 0% before the mid 1980s to more than 80% by 2007 in the United States [8]. To decipher how E. faecium has become a globally disseminated nosocomial pathogen, several whole-genome sequencing-based studies have been performed [9,10,11]. Phylogenetic analysis based on whole-genome sequencing has shown that the E. faecium population is formed by two clades: clade A contains mainly clinical or hospital-associated isolates, while clade B contains primarily commensal/community-associated isolates [12, 13]. Further work has shown that a second split occurred within clade A approximately 75 years ago, resulting in two subclades, subclade A1 and subclade A2. Subclade A1 contains mainly the clinical isolates, including the successful hospital-adapted sequence types of the clonal complex 17 (CC17) genogroup defined by multilocus sequence ty**, while subclade A2 mostly consisted of the animal-associated isolates [9]. However, a recent comparative genomic study of health-care associated E. faecium from UK and Ireland did not support the subdivision of clade A into the two subclades [11]. Most published studies have so far focused on isolates from clinical settings and animal origins. In fact, E. faecium is ubiquitous in nature and has been isolated from a wide range of environments [14]. Thus, to gain deeper insights into the evolutionary origin of E. faecium, a large sampling from multiple-habitats is required to better represent the natural distribution of this microbial group.
Apart from human/animal GI tracts and clinical samples, fermented dairy foods are indeed natural habitats for E. faecium [15, 16]. Isolates of E. faecium originated from fermented dairy products do not only act as bystanders but also participate in the biochemical reactions that occur during the ripening process [17]. Some E. faecium isolates are known to confer beneficial effects to the host and are even marketed as commercial probiotics (e.g. E. faecium isolate T110). A recent comparative genome analysis has revealed genetic features of potential probiotic E. faecium isolates [18]. Providing the probiotic, non-pathogenic non-probiotic, and pathogenic nature among the E. faecium isolates, it is therefore interesting to investigate if there are links between their isolation environment, physiological features, and phylogeny.
Another interesting question is if the dairy isolates are a product of faecal contamination or environmental habitat, since the selective pressure for E. faecium to survive in dairy products is enormously different from the complex niches within human and animal hosts. The dairy has relative simple carbon and nitrogen source, convergent evolution is likely to be dominant when the bacteria adapt to the dairy environment. Thus, comparative study integrating environmental isolates such as dairy isolates as reference could provide insights to understand not only of the hospital-acquired infection and multidrug resistance of E. faecium but also their mechanisms of environmental adaptation.
To gain deeper insights into the genetic relationships of the dairy isolates and the isolates from human and animals, this study performed a comparative genomic analysis on 161 E. faecium, of which 54 dairy isolates were isolated and sequenced by our laboratory while the other 107 genome sequences were retrieved from a public genome database.
Results
General characteristics of the genomes of E. faecium
In this study, comparative genomic analysis was conducted on 161 isolates, including 33 isolates from human GI, 39 isolates from human blood, nine isolates from human UT, eight isolates from chicken GI, two isolates from dog GI, seven isolates from pig GI, one isolate from Mammuthus (M.) primigenius GI, 61 isolates from dairy products, and one isolate from soy, respectively. The E. faecium genomes had a low overall G + C content ranging from 37.4 to 38.6% (Additional file 1). The average genome size was 2.85 ± 0.20 Mb, with 2,765 ± 187 predicted genes (Additional file 1). Furthermore, apart from the two dog GI-originated isolates, significant differences existed in the genome size and/or the number of predicted genes between isolates from different environments (Fig. 1). Generally, the human-originated isolates had the largest mean genome size and the maximum mean number of predicted genes, which were significantly higher than the isolates of pig GI (P < 0.01), chicken GI (P < 0.01), and dairy (P < 0.01). Meanwhile, it is interesting to note that although no significant difference was observed in the genome size among these three groups, significantly more predicted genes were identified in the dairy isolates compared with those originated from pig GI (P = 0.006) and chicken GI (P = 0.048). Further analysis showed that the dairy isolates had more short genes (100–200 bp) than pig and chicken isolates (Additional file 2). On average, the dairy isolates had 150 fewer predicted genes compared with those of human origin. The missing genes represented mainly the COG functional categories of replication, recombination and repair [L] and carbohydrate transport and metabolism [G] (Additional file 3). Since the sample size of both the M. primigenious GI and soy groups was too small (n = 1 in each case), no statistical analysis could be performed.

Genome size (a) and number of predicted genes (b) of Enterococcus faecium isolates from different environments. The asterisk (*) and double asterisks (**) represent p < 0.05 and < 0.01 from the one-way ANOVA test, respectively
To evaluate the genetic difference between strains, we generated an average nucleotide identity (ANI) plot (Fig. 2). The ANI values across all strains ranged from 94.23 to 99.99, with the lowest ANI value between the isolates E3548 (human blood) and TW5–3 (dairy). The two dairy isolates, had the highest ANI (99.99). The ANI values of the dairy and human isolates ranged from 94.53 to 99.99 and 94.27 to 99.99, respectively. The genetic closeness between strains could be classed into two groups according to the ANI value (20 isolates with a lower overall ANI value, while the other 141 isolates had a higher overall ANI), although no obvious grou** pattern was noted based on the bacterial isolation source (Fig. 2).

The heatmap of the average nucleotide identity of 161 Enterococcus faecium isolates
The pan- and core-genome of E. faecium
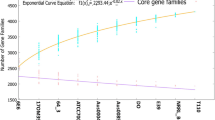
The pan-genome of the 161 E. faecium isolates comprised 12,457 gene families, and the pan-genome size increased with the number of genomes (Fig. 3a). In contrast, the core-genome size gradually decreased with the addition of deciphered genomes. When the number of genomes reached 130–140, the core-genome size remained stable (Fig. 3b). The core gene set contained 1013 genes, corresponding to 36.6% of the average number of predicted genes per genome (2765 genes). In other words, almost two-thirds of the predicted genes in each genome were accessory.

Pan-genomes (a) and core-genomes (b) of the species Enterococcus faecium. The gene accumulation curves describe the number of genes obtained by adding a new genome to a previous set. The procedure was repeated 1,000 times by randomly modifying the order of integration of genomes
The results of the functional analysis of the pan- and core-genome are shown in Table 1. Around one-third (33.4%) of the core genes were distributed to the COG functional categories of translation, ribosomal structure and biogenesis [J]; amino acid transport and metabolism [E]; transcription [K]; and carbohydrate transport and metabolism [G]. The pan genes mainly fell into categories representing the COG categories: carbohydrate transport and metabolism [G]; cell wall/membrane/envelope biogenesis [M]; replication, recombination and repair [L]; and transcription [K]. The COG category genes of carbohydrate transport and metabolism [G] were enriched in both the pan- and core-genomes. However, it was the most abundant COG category class (12.74%) detected only within the pan-genome but not the core-genome (7.36%), and it resulted in a large pan-genome expansion of 9.72-fold, suggesting strong selective pressure in these genes and niche selection.
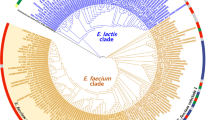
Phylogenetic reconstruction of E. faecium
A phylogenetic tree was constructed to investigate the evolutionary relationship among the studied isolates (Fig. 4, Additional file 4). Here three isolates of E. mundtii, the closest phylogenetic relatives of E. faecium, were included as outgroups. The topology of the phylogenetic tree revealed two clades, which were defined as clades I and II in this study (Fig. 4). Furthermore, two subclades could be identified within clade I, defined as subclades IA and IB. Subclade IA was larger and comprised mostly the human isolates (64 of the 67 isolates), as well as the two isolates from dog GI and one isolate from a dairy product of ** the E. faecium genomes, the environment seems to play an important role too. Moreover, the large genome plasticity of E. faecium could contribute to the high versatility of this species in adapting successfully to a wide range of environments from dairy to harsh conditions such as the hospital environment and GI [23].
Finally, the increasing medical significance of the health-care associated E. faecium lineages has driven us to analyse the spectrum of antibiotic resistance genes in these genomes in detail. Cluster analysis based on the distribution of vancomycin resistance genes found three types of antibiotic resistance spectrum: vanA, vanB, and van absent (Fig. 5). The dairy isolates had the fewest antibiotic resistance genes and belonged to the van absent category, making them significantly different from the human and animal isolates. Such results also support that the dairy isolates were not derived merely from faecal contamination but existed as independent lineages. Antibiotic resistance has been acquired and disseminated throughout enterococci via horizontal transfer of mobile genetic elements [24]. The antibiotic resistance genes render human and animal isolates successful colonizers or even invaders of human and animal GI tracts [5], while the antibiotic selective pressure is much less in dairy products, resulting in a general absence of this spectrum of genes among the dairy isolates. Among the other two types of antibiotic resistance profile, the vanA type cluster is the most frequently encountered type of glycopeptide resistance in enterococci; and isolates that have acquired vanA are resistant to high levels of vancomycin [25]. The organization and functionality of the vanB cluster is similar to that of vanA, but the resistant levels are variable [25]. In this study, about three quarters of isolates from human and animals carried vancomycin resistance genes, which is more prevalent than E. faecalis [26].
Conclusions
In conclusion, the comparative genomic study of E. faecium showed significant differences between isolates from dairy products and humans. The dairy isolates existed as independent lineages rather than a product of faecal contamination based on our phylogenetic analysis. Results from the phylogenetic and genome-wide association studies together revealed links between the bacterial isolation environment, physiological features, and phylogeny. Moreover, the great genomic plasticity of E. faecium probably provides the strong environmental adaptability to enable the successful adaptation of this species to various ecological niches.
Methods
Bacterial isolates and genomes
A total of 161 genomes of E. faecium were subjected to whole genome sequencing-based analysis. Among them, 54 isolates were previously isolated from naturally fermented dairy products in China and Russia from 2007 to 2014 by our laboratory; they were firstly sequenced in this study (Additional file 1). The other E. faecium genomes were retrieved from the Genbank database on April, 2017. At the start of this work, all accessible genome records of E. faecium were investigated to include only those having clear documentation of bacterial isolation source and of high sequencing quality. These two criteria were important to ensure comparability between genomes and grou** of isolation environment. After screening, 107 genomes remained, which were from human GI (33 genomes), human blood (39 genomes), human UT (9 genomes), chicken GI (8 genomes), dog GI (2 genomes), pig GI (7 genomes), M. primigenius GI (1 genome), dairy products (7 genomes), and soy (1 genome), respectively (Additional file 1).
The genomes of three E. mundtii isolates (accession numbers: AP013036, CP018061, and CP022340) were downloaded and included as outgroups in the phylogenetic tree reconstruction.
DNA extraction
Bacteria were grown in de Man Rogosa and Sharpe (MRS) broth under anaerobic conditions at 37 °C. Bacterial DNA was extracted with a commercial DNA extraction kit (OMEGA D3350–02) according to the manufacturer’s instructions. The amount of extracted genomic DNA was quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA). Fragment libraries (200 to 300 bp) were constructed only with high-quality DNA (OD260/280 = 1.8~2.0, > 6 μg).
Whole-genome sequencing, assembly, and annotation
The whole-genome sequencing was done using the Illumina MiSeq platform (Illumina Inc. U.S.A) by generating 2 × 150 bp paired-end libraries using the Nextera DNA Sample Preparation Kit (Illumina Inc., U.S.A) following the manufacturer’s instructions. On average, 1,576 Mb of high-quality data were generated for each isolate, corresponding to 395- to 969-fold sequencing depth (Additional file 1).
The paired-end reads were first assembled de novo using SOAPdenovo v1.06 [27]. GapCloser (http://sourceforge.net/projects/soapdenovo2/files/GapCloser/) was used to fill local inner gaps and correct single base errors. Glimmer v3.02 was used to predict putative coding sequences [28]. RAST 2.0 [29] and COG database [30] were used to annotate the functions of predicted open reading frames (ORFs). The assembled genomes of the 54 isolates were deposited in the National Center for Biotechnology Information GenBank database under the accession numbers of PGPI00000000 to PGTQ00000000 (Additional file 1).
Construction of core- and pan-genomes
All predicted ORFs were firstly assigned to the respective gene families. Then, the core- and pan-genomes of E. faecium were constructed using the SiLiX software [31] on the basis of homologous gene families. Two ORFs would be assigned to the same family if the amino acid sequence identity was greater than 80% and if the shorter ORF aligned over 80% of that of the longer one. The pan-genome comprised the total number of non-redundant gene families within the complete dataset, while the core-genome included gene families which were present in all E. faecium genomes. The longest ORF from each gene family was used as the representative sequence for functional annotation and phylogenetic analysis.
Construction of phylogenetic tree
The phylogenetic tree was constructed using the core genes of 161 E. faecium and three E. mundtii isolates. The species E. mundtii was the closest relative of E. faecium and was thus used as outgroups. Briefly, the nucleotide sequences of the core genes were aligned using MUSCLE v3.8.31 [32], and the unreliable alignment regions and intragenic homologous recombination were removed by using Gblocks (http://molevol.cmima.csic.es/castresana/Gblocks.html) and Gubbins (http://www.sanger.ac.uk/science/tools/gubbins) [33], respectively. A maximum likelihood tree based on the concatenated alignments was built using FastTree 2.1.8 [34] with 1,000 bootstrap iterations. The phylogenetic tree was visualized with the online tool, Interactive Tree Of Life (iTOL) [35].
Identification of environment-specific genes
In order to identify environment-specific genes, the portion of variable genes found in the pan-genome was analysed to determine if there was significant association between gene distribution and bacterial isolation source. Environment-specific genes were defined as those having a significantly higher prevalence in isolates from one particular isolation source compared with the overall occurrence across all genomes. Scoary 1.6.16 was applied to identify environment-specific genes based on 1,000 permutation replicates. The results were corrected for multiple testing with Benjamini-Hochberg correction. A corrected P-value < 0.05 was considered significant [36].
Identification of antibiotic resistance genes
A BLASTp search was conducted with the predicted genes from all investigated genomes against the CARD (Comprehensive Antibiotic Resistance Database; http://arpcard.mcmaster.ca) to identify potential antibiotic resistance genes (E-value <1e-15, coverage > 90% and sequence identity > 85%) [37, 38]. The hierarchical cluster analysis was based on the presence or absence of the antibiotic resistant genes and the heatmap was drawn using the “pheatmap” package of the R software (V3.6.0).
Identification of virulence factors
A BLASTp search was done with the predicted genes from all investigated genomes against the VFDB (Virulence Factor Database) to identify genes related to potential virulence factors (E-value <1e-15, coverage > 90% and sequence identity > 85%) [38, 39].
Statistical analysis
Data are presented as means ± SEM. One-way ANOVA followed by Tukey’s post-hoc test was used for statistical significance determination using the SPSS Statistics 19 (IBM, Armonk, New York, USA). Significance was set at P < 0.05.
Availability of data and materials
The individual genome assemblies of 54 isolates were deposited in the National Center for Biotechnology Information under accession numbers PGPI00000000 to PGTQ00000000 (Additional file 1).
Abbreviations
- E :
-
Enterococcus
- GI:
-
Gastrointestinal
- M.:
-
Mammuthus
- MRS:
-
Man Rogosa and Sharpe
- ORFs:
-
Open reading frames
- UT:
-
Urinary tract
References
Murray BE. The life and times of the Enterococcus. Clin Microbiol Rev. 1990;3(1):46–65.
Arias CA, Murray BE. The rise of the Enterococcus: beyond vancomycin resistance. Nat Rev Microbiol. 2012;10(4):266–78.
Gilmore MS, Lebreton F, van Schaik W. Genomic transition of enterococci from gut commensals to leading causes of multidrug-resistant hospital infection in the antibiotic era. Curr Opin Microbiol. 2013;16(1):10–6.
Guzman Prieto AM, van Schaik W, Rogers MR, Coque TM, Baquero F, Corander J, Willems RJ. Global emergence and dissemination of enterococci as nosocomial pathogens: attack of the clones? Front Microbiol. 2016;7:788.
Gao W, Howden BP, Stinear TP. Evolution of virulence in Enterococcus faecium, a hospital-adapted opportunistic pathogen. Curr Opin Microbiol. 2017;41:76–82.
Willems RJ, van Schaik W. Transition of Enterococcus faecium from commensal organism to nosocomial pathogen. Future Microbiol. 2009;4(9):1125–35.
Moscoso M, Domenech M, Garcia E. Vancomycin tolerance in Gram-positive cocci. Environ Microbiol Rep. 2011;3(6):640–50.
Arias CA, Murray BE. Emergence and management of drug-resistant enterococcal infections. Expert Rev Anti Infect Ther. 2008;6(5):637–55.
Lebreton F, van Schaik W, McGuire AM, Godfrey P, Griggs A, Mazumdar V, Corander J, Cheng L, Saif S, Young S et al: Emergence of epidemic multidrug-resistant Enterococcus faecium from animal and commensal strains. mBio 2013, 4(4).
Lebreton F, Manson AL, Saavedra JT, Straub TJ, Earl AM, Gilmore MS. Tracing the enterococci from Paleozoic origins to the hospital. Cell. 2017;169(5):849–61 e813.
Raven KE, Reuter S, Reynolds R, Brodrick HJ, Russell JE, Torok ME, Parkhill J, Peacock SJ. A decade of genomic history for healthcare-associated Enterococcus faecium in the United Kingdom and Ireland. Genome Res. 2016;26(10):1388–96.
Galloway-Pena J, Roh JH, Latorre M, Qin X, Murray BE. Genomic and SNP analyses demonstrate a distant separation of the hospital and community-associated clades of Enterococcus faecium. PLoS One. 2012;7(1):e30187.
Palmer KL, Godfrey P, Griggs A, Kos VN, Zucker J, Desjardins C, Cerqueira G, Gevers D, Walker S, Wortman J et al: Comparative genomics of enterococci: variation in Enterococcus faecalis, clade structure in E. faecium, and defining characteristics of E. gallinarum and E. casseliflavus. mBio 2012, 3(1):e00318–00311.
McGowan LL, Jackson CR, Barrett JB, Hiott LM, Fedorka-Cray PJ. Prevalence and antimicrobial resistance of enterococci isolated from retail fruits, vegetables, and meats. J Food Prot. 2006;69(12):2976–82.
Lebreton F, Willems RJL, Gilmore MS: Enterococcus Diversity, Origins in Nature, and Gut Colonization. In: Enterococci: From Commensals to Leading Causes of Drug Resistant Infection. Edited by Gilmore MS, Clewell DB, Ike Y, Shankar N. Boston; 2014.
Mannu L, Paba A, Daga E, Comunian R, Zanetti S, Dupre I, Sechi LA. Comparison of the incidence of virulence determinants and antibiotic resistance between Enterococcus faecium strains of dairy, animal and clinical origin. Int J Food Microbiol. 2003;88(2–3):291–304.
Foulquie Moreno MR, Sarantinopoulos P, Tsakalidou E, De Vuyst L. The role and application of enterococci in food and health. Int J Food Microbiol. 2006;106(1):1–24.
Ghattargi VC, Gaikwad MA, Meti BS, Nimonkar YS, Dixit K, Prakash O, Shouche YS, Pawar SP, Dhotre DP. Comparative genome analysis reveals key genetic factors associated with probiotic property in Enterococcus faecium strains. BMC Genomics. 2018;19(1):652.
Costa Y, Galimand M, Leclercq R, Duval J, Courvalin P. Characterization of the chromosomal aac (6′)-ii gene specific for Enterococcus faecium. Antimicrob Agents Chemother. 1993;37(9):1896–903.
Draker KA, Northrop DB, Wright GD. Kinetic mechanism of the GCN5-related chromosomal aminoglycoside acetyltransferase AAC (6′)-ii from Enterococcus faecium: evidence of dimer subunit cooperativity. Biochemistry. 2003;42(21):6565–74.
Ogier JC, Serror P. Safety assessment of dairy microorganisms: the Enterococcus genus. Int J Food Microbiol. 2008;126(3):291–301.
Goncharov A, Grigorjev S, Karaseva A, Kolodzhieva V, Azarov D, Akhremenko Y, Tarasova L, Tikhonov A, Masharskiy A, Zueva L et al: Draft Genome Sequence of Enterococcus faecium Strain 58m, Isolated from Intestinal Tract Content of a Woolly Mammoth, Mammuthus primigenius. Genome announcements 2016, 4 (1).
Cattoir V, Giard JC. Antibiotic resistance in Enterococcus faecium clinical isolates. Expert Rev Anti-Infect Ther. 2014;12(2):239–48.
Palmer KL, Kos VN, Gilmore MS. Horizontal gene transfer and the genomics of enterococcal antibiotic resistance. Curr Opin Microbiol. 2010;13(5):632–9.
Courvalin P. Vancomycin resistance in gram-positive cocci. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2006;42(Suppl 1):S25–34.
He Q, Hou Q, Wang Y, Li J, Li W, Kwok LY, Sun Z, Zhang H, Zhong Z. Comparative genomic analysis of Enterococcus faecalis: insights into their environmental adaptations. BMC Genomics. 2018;19(1):527.
Li R, Zhu H, Ruan J, Qian W, Fang X, Shi Z, Li Y, Li S, Shan G, Kristiansen K, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010;20(2):265–72.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics. 2007;23(6):673–9.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC bioinformatics. 2003;4:41.
Miele V, Penel S, Duret L. Ultra-fast sequence clustering from similarity networks with SiLiX. BMC bioinformatics. 2011;12:116.
Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004;5:113.
Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR: Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 2015, 43(3).
Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol. 2009;26(7):1641–50.
Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2019.
Brynildsrud O, Bohlin J, Scheffer L, Eldholm V. Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 2016;17(1):238.
McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, et al. The comprehensive antibiotic resistance database. Antimicrob Agents Chemother. 2013;57(7):3348–57.
Tang F, Wang J, Li D, Gao S, Ren J, Ma L, Liu F, Zhuge X, Yan G, Lu Y, et al. Comparative genomic analysis of 127 Escherichia coli strains isolated from domestic animals with diarrhea in China. BMC Genomics. 2019;20(1):212.
Chen L, Yang J, Yu J, Yao Z, Sun L, Shen Y, ** Q. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33(Database issue):D325–8.
Acknowledgements
Not applicable.
Funding
This research was supported by the National Natural Science Foundations of China (Grant No. 31601451, 31622043, and 31430066) and the China Agriculture Research System (Grant CARS-36). The funding bodies had no role in the design of the study or collection, analysis, and interpretation of data or in writing the manuscript.
Author information
Authors and Affiliations
Contributions
ZZ, HZ, and ZS designed the study. ZZ, QH, YS, and WL performed the de novo assembly and comparative genomics analyses. ZZ and LK wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
List of the Enterococcus faecium isolates analysed in this study. (XLSX 25 kb)
Additional file 2:
The length distribution of coding DNA sequences (CDSs) in Enterococcus faecium genomes. (XLSX 62 kb)
Additional file 3:
Clusters of Orthologous Groups (COGs) functional categories of predicted genes of dairy and human isolates. [J] Translation, ribosomal structure and biogenesis; [K] Transcription; [L] Replication, recombination and repair; [D] Cell cycle control, cell division, chromosome partitioning; [V] Defense mechanisms; [T] Signal transduction mechanisms; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [U] Intracellular trafficking, secretion, and vesicular transport; [O] Post-translational modification, protein turnover, and chaperones; [C] Energy production and conversion; [G] Carbohydrate transport and metabolism; [E] Amino acid transport and metabolism; [F] Nucleotide transport and metabolism; [H] Coenzyme transport and metabolism; [I] Lipid transport and metabolism; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport, and catabolism; [R] General function prediction only; [S] Function unknown. (PDF 208 kb)
Additional file 4:
Phylogenetic tree constructed based on the core genes of Enterococcus faecium isolates. Bootstrap values are shown at the nodes. (PDF 35 kb)
Additional file 5:
The antibiotic resistance genes in the 161 isolates of Enterococcus faecium (XLSX 71 kb)
Additional file 6:
The virulence factors in the 161 isolates of Enterococcus faecium (XLSX 32 kb)
Additional file 7:
List of environment-specific genes (XLSX 30 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhong, Z., Kwok, LY., Hou, Q. et al. Comparative genomic analysis revealed great plasticity and environmental adaptation of the genomes of Enterococcus faecium. BMC Genomics 20, 602 (2019). https://doi.org/10.1186/s12864-019-5975-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5975-8