
Abstract
Background
Bovine viral diarrhea virus (BVDV) is an economically important viral pathogen of domestic and wild ruminants. Apart from cattle, small ruminants (goats and sheep) are also the susceptible hosts for BVDV. BVDV infection could interfere both of the innate and adaptive immunity of the host, while the genes and mechanisms responsible for these effects have not yet been fully understood. Peripheral blood mononuclear cells (PBMCs) play a pivotal role in the immune responses to viral infection, and these cells were the target of BVDV infection. In the present study, the transcriptome of goat peripheral blood mononuclear cells (PBMCs) infected with BVDV-2 was explored by using RNA-Seq technology.
Results
Goat PBMCs were successfully infected by BVDV-2, as determined by RT-PCR and quantitative real-time RT-PCR (qRT-PCR). RNA-Seq analysis results at 12 h post-infection (hpi) revealed 499 differentially expressed genes (DEGs, fold-change ≥ ± 2, p < 0.05) between infected and mock-infected PBMCs. Of these genes, 97 were up-regulated and the remaining 352 genes were down-regulated. The identified DEGs were found to be significantly enriched for locomotion/ localization, immune response, inflammatory response, defense response, regulation of cytokine production, etc., under GO enrichment analysis. Cytokine-cytokine receptor interaction, TNF signaling pathway, chemokine signaling pathway, etc., were found to be significantly enriched in KEGG pathway database. Protein-protein interaction (PPI) network analysis indicated most of the DEGs related to innate or adaptive immune responses, inflammatory response, and cytokine/chemokine-mediated signaling pathway. TNF, IL-6, IL-10, IL-12B, GM-CSF, ICAM1, EDN1, CCL5, CCL20, CXCL10, CCL2, MAPK11, MAPK13, CSF1R and LRRK1 were located in the core of the network and highly connected with other DGEs.
Conclusions
BVDV-2 infection of goat PBMCs causes the transcription changes of a series of DEGs related to host immune responses, including inflammation, defense response, cell locomotion, cytokine/chemokine-mediated signaling, etc. The results will be useful for exploring and further understanding the host responses to BVDV-2 infection in goats.
Similar content being viewed by others


Background
Bovine viral diarrhea virus (BVDV) is the prototypic member of the genus Pestivirus in the family Flaviviridae, and two main different BVDV species have been recognized as BVDV-1 and BVDV-2 [1]. BVDV infection decreases productive performance and causes considerable economic losses in cattle industry worldwide [2]. Infections with both species of BVDV can induce similar diseases, from subclinical infections to severe clinical diseases including acute diarrhea, respiratory diseases, reproductive failures, congenital defects, and increased mortality due to immunosuppression [2,3,4]. Persistent infection is the common type of infection in cattle and the persistently infected (PI) animals are considered the main source of BVDV transmission [2, 5]. PI animals had also been detected in heterologous species, which amplify and facilitate the reservoirs for BVDV [6, 7]. Evidence of BVDV infection exists in 7 families (over 50 species) of Artiodactyla including Antilocapridae, Bovidae, Camelidae, Cervidae, Giraffidae, Suidae, and Tragulidae [6]. The circulation of BVDV-1 and BVDV-2 in cattle, pigs had been identified in China [8,9,10]. Our previous study has identified the prevalence of BVDV-1 in Chinese goat herds [11]. The infection of BVDV-2 in goat or sheep has been confirmed in India, Korea [12,13,14]. Therefore, the risk and prevalence of BVDV-1 and BVDV-2 in goat/sheep herds needs urgent attention.
Recently, high-throughput RNA-Sequencing (RNA-Seq) technologies give the opportunity to produce large numbers of sequence data in non-model organisms, and this method is better than the traditional microarray analysis [15, 16]. It provides a thorough understanding of the host defense mechanisms and immune evasion strategies of viral infection [Full size table
DEGs analysis and functional annotation
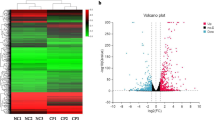
After the gene map** and the Cuffdiff analyses in terms of FRKM, a total of 449 genes were identified as significantly differentially expressed for infected group (B2), when comparing with the mock-infected control group (N, fold change (FC) ≥ ± 2, p < 0.05). Among the 449 genes, 97 were up-regulated and 352 genes were down-regulated (Fig. 2, Additional file 4: Figure S1, Additional file 1: Table S1).

Summary of the differentially expressed genes between mock and BVDV-2 infected samples at 12hpi
The 449 DEGs were annotated to 54 different GO terms. The up-regulated DEGs were annotated to 38 GO terms and the down-regulated DEGs were annotated to 53 GO terms (Fig. 3). The most annotated GO terms were metabolic process (BP), cellular process (BP), response to stimulus (BP), biological regulation (BP), localization (BP), cell (CC), cell part (CC), membrane (CC), membrane part (CC), extracellular region (CC), organelle (CC) and binding (MF), etc. (Fig. 3).

GO annotation for the DEGs between mock and BVDV-2 infected goat PBMCs
Functional and PPI analysis of DEGs
After GO enrichment analysis, DEGs were enriched into different GO terms. For immune related DEGs, significant enrichment was observed in regulation of locomotion/ localization, immune response, inflammatory response, immune system process, defense response, regulation of cytokine production of BP group and in cytokine activity, chemokine activity, receptor binding of MF group. (Fig. 4 and Additional file 2: Table S2).

GO enrichment analysis for the DEGs between mock and BVDV-2 infected goat PBMCs. Circles indicate numbers of enriched genes and colors depict the P value
To further define DEGs function, KEGG pathway/enrichment analysis was performed. Among the fifteen significantly enriched pathways, cytokine-cytokine receptor interaction, TNF signaling pathway, chemokine signaling pathway, complement and coagulation cascades and NOD-like receptor signaling pathway were found to be enriched to canonical pathways (Fig. 5, Table 2 and Additional file 3: Table S3). The cytokine-cytokine receptor interaction pathway was the pathway enriched with most number of DEGs (n = 29). For the 29 DEGs, CCL4, CCL3, CXCL10, CCL5, CCL22, CCL20, GM-CSF, TNF, IL-6, IL-17A, IL-12B, IL-19, IL-10, TNFRSF13C, TNFRSF8, TNFRSF9 and XCL1 were up-regulated; while TNFSF12, CSF1R, TNFRSF21, CSF3R, regakine-1(LOC102170772), CCL2, CCL24, CCL17, CCL14, CCL25, IL-5RA and PPBP were down-regulated. In addition, TNF, IL-6, CXCL10, CCL4, CCL3, CCL5, CCL20, CCL2 and regakine-1 were enriched in at least three of the six pathways mentioned above (Table 2).

Top 30 pathways enriched in KEGG database for the DEGs between mock and BVDV-2 infected goat PBMCs. Circles indicate numbers of enriched genes and colors depict the P value
STRING analysis was used to explore the potential interaction network of the DEGs. As shown in Fig. 6, most of the DEGs related to innate or adaptive immune responses, inflammatory response, cytokine/chemokine-mediated signaling pathway, etc. Among the up-regulated genes, TNF, IL-6, IL-10, IL-12B, GM-CSF, ICAM1, EDN1, CCL20, CXCL10 and CCL5 were located in the core of the network and linked to lots of other DGEs; for the down-regulated genes, the key points included CCL2, MAPK11, MAPK13, CSF1R and LRRK1, etc., which linked to more genes. In addition, not all DEGs showed connection with others because their functions were either unrelated or have not yet been clarified (Additional file 5: Figure S2). These DGEs were not included in Fig. 6 during the analysis.

PPI network of the selected DEGs based on STING analysis results and fold change information. The up-regulated genes are shown in red and down-regulated genes are shown in blue with the gradient showing the extent of expression. The size of the node indicates connectivity
Partial validation of RNA-Seq data
To further validate the RNA-Seq data, DEGs with annotations associated with immune responses from RNA-seq were selected for qRT-PCR analysis. As shown in Table 3, eighteen selected genes exhibited a concordant direction both in RNA-Seq and qRT-PCR analysis. The correlation coefficient between RNA-Seq and qRT-PCR results was high (R2 = 0.91). Some of the DEGs were further determined by Western blot or ELISA, as shown in Fig. 7, the expression of Annexin A2 decreased obviously (Fig. 7a) and the expression of TNF-α, GM-CSF and IL-6 were increased significantly (Fig. 7b). In addition, Viperin (used as a control) expression showed no change between infected and mock-infected group, which was consistent with the RNA-Seq result. These results confirmed that the differential expression genes identified by RNA-Seq is reliable.

Partial validation of RNA-Seq data by Western blot and ELISA. a The expression of Annexin A2 and Viperin in the samples was determined by Western blot using rabbit anti-Viperin polyclonal antibody (Abcam) and mouse anti-AnnexinA2 antibody (Santa Cruz). b The concentration of GM-CSF, TNF- and IL-6 present in the samples were determined by commercial ELISA kit and calculated with the formula derived from the standard curve. Data was shown as the mean ± S.D. Columns marked with * (P < 0.05) are significantly different from each other

{kind=link}