
Abstract
Background
Technological advances have enabled transcriptome characterization of cell types at the single-cell level providing new biological insights. New methods that enable simple yet high-throughput single-cell expression profiling are highly desirable.
Results
Here we report a novel nanowell-based single-cell RNA sequencing system, ICELL8, which enables processing of thousands of cells per sample. The system employs a 5,184-nanowell-containing microchip to capture ~1,300 single cells and process them. Each nanowell contains preprinted oligonucleotides encoding poly-d(T), a unique well barcode, and a unique molecular identifier. The ICELL8 system uses imaging software to identify nanowells containing viable single cells and only wells with single cells are processed into sequencing libraries. Here, we report the performance and utility of ICELL8 using samples of increasing complexity from cultured cells to mouse solid tissue samples. Our assessment of the system to discriminate between mixed human and mouse cells showed that ICELL8 has a low cell multiplet rate (< 3%) and low cross-cell contamination. We characterized single-cell transcriptomes of more than a thousand cultured human and mouse cells as well as 468 mouse pancreatic islets cells. We were able to identify distinct cell types in pancreatic islets, including alpha, beta, delta and gamma cells.
Conclusions
Overall, ICELL8 provides efficient and cost-effective single-cell expression profiling of thousands of cells, allowing researchers to decipher single-cell transcriptomes within complex biological samples.
Similar content being viewed by others
Background
Single-cell RNA sequencing (RNA-seq) has rapidly evolved over the last few years, providing new understanding of cell composition and identity in normal and diseased settings [1, 2]. Transcriptional profiling at the single-cell level facilitates identification of new cell types and understanding of cellular heterogeneity, aids in lineage tracing, and elucidates hierarchical relationships among cell types in the course of development or disease progression [2,3,4,5,6,7]. For example, based on their transcriptome patterns, more than 40 subtypes of neurons were identified in the mouse cortex [7, 8]. Single-cell RNA-seq also identified a rare cell type in colon tissue [4].
Initial transcriptional profiling at the single-cell level was done using fluorescence-activated cell sorting (FACS). However, FACS-based techniques have limited throughput and are not cost-effective. Recently, methods that enable single-cell transcriptome and whole-genome sequencing without the need for FACS have been reported. Notably, microfluidic-based single-cell isolation (e.g. Fluidigm C1) and transcriptome analysis was performed successfully, though the throughput and the ability to capture a large range of cell sizes is limiting [6]. The C1-based method does not employ molecular barcodes for eliminating PCR duplicates during cDNA conversion and can potentially lead to data bias [6, 9]. Recently, encapsulation of thousands of cells using droplet-based microfluidic methods for single-cell RNA-seq have been demonstrated [10, 11]. While droplet-based technology is encouraging, further development of the experimental technique and analysis tools are needed for its wider adoption [9, 12, 13].
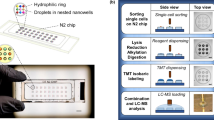
Nanowell-based deposition of single cells is another promising approach for characterizing single-cell transcriptomes [14]. In this study we assessed the performance and utility of the ICELL8 system for massively parallel single-cell gene expression profiling (Fig. 1a). The ICELL8 system uses a multi-sample nanodispenser (MSND) to dispense single cells into a 5184-nanowell microchip. Imaging software is used to select single-cell-containing wells that are then processed to obtain single-cell transcriptome data. We showed that the system has a low rate of cell multiplets and high single-cell purity. In addition, we characterized transcriptomes of more than a thousand cultured cells and were able to identify representative cell subtypes from a complex tissue sample.

Overview of the ICELL8 single-cell RNA-seq workflow. a High-throughput single-cell RNA-seq is performed by dispensing a single-cell suspension onto a microchip (containing 5184 nanowells), followed by microchip imaging, and on-chip cDNA generation. The process is completed by standard in-tube NGS library generation, Illumina sequencing and computational data analysis. b Histogram of the number of wells containing 0, 1, 2, 3 or 4 cells as determined by image analysis software. Boxes and error bars indicated the median and the range, respectively, for several microchips (n = 5). c Schematic illustration of sequencing library constructs and sequence read position and length
Results
Microchip for single-cell isolation
The ICELL8 microchip is made of aluminum alloy (41 mm2) and contains 5184 nanowells arranged in a square layout (72 × 72 wells). Each nanowell holds 150 nl and contains preprinted oligonucleotides; each oligonucleotide includes an oligo-(dT30) primer, a well-specific sequence (11 bp) used for cell barcoding and a unique molecular identifier (UMI, 10 bp; Fig. 1). The cell barcode is used to identify cDNA molecules generated from an individual cell, while UMIs identify individual mRNA molecules [15]. A similar microchip-based technology has been used previously in targeted sequencing applications [16, 17].
Cell suspensions were fluorescently labeled with live/dead stain (Hoechst 33324/Propidium Iodide, see Methods) prior to their dispensing into the microchip nanowells using the MSND (Additional file 1: Figure S1). The MSND is an 8-channel microsolenoid controlled dispenser that delivers ≥30 nl volumes using non-contact dispensing. The MSND dispenses up to eight different samples (one sample per channel) into a single microchip in approximately 12 minutes. To minimize evaporation, the microchip is enclosed in a controlled humidity and temperature chamber. Cross contamination between nanowells due to dispense tip misalignment was assessed using a checkerboard assay (see Methods, Additional file 2: Figure S2). In a test involving 11 MSND instruments we observed the average percentage of wells affected by misalignment to be 0.08%. Cells are dispensed by a limiting dilution; assuming a Poisson distribution for the number of cells per well, about one third of the 5184 nanowells contain a single cell under optimal conditions.
Following cell dispensing, the microchip was centrifuged to collect cells in a single plane and then imaged using a standard microscope with an automatic stage, a 4× objective and a charge-coupled device (CCD) camera. After imaging, the microchip was sealed and stored at −80 °C until ready for use (Additional file 1: Figure S1). Next we used imaging software (CellSelect, see Materials and Methods) to automatically and/or manually identify wells that contain single cells (Fig. 1b). A file containing positional information on identified candidate wells (dispense file) was then used to selectively deliver reverse transcriptase (RT) master mix to designated wells.
Single-cell barcoding and sequencing
Cells in the microchip were lysed by freeze-thaw (see Methods). Under the directions from the dispense file, the MSND dispensed RT master mix into selected wells and cDNA synthesis was performed in those wells using the Single Cell Barcoding and Sequencing method (SCRB-seq) [18]. cDNAs from hundreds of cells were then pooled into a single tube, purified and amplified by standard practices. The amplified cDNAs were subjected to transposon-mediated fragmentation (“tagmentation”), PCR amplified and converted to an Illumina-compatible NGS library (Additional file 1: Figure S1). RNA-seq libraries were sequenced using paired-end sequencing where read 1 (25 bp) contained the well barcode and UMI and read 2 (50 bp) captured the cDNA sequence (Fig. 1c).
Sequencing data processing and quality control
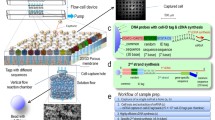
To analyze sequencing reads generated from pooled single-cell RNA-seq libraries we developed a computational pipeline shown in Fig. 2a. Reads from different wells were demultiplexed based on a perfect match to the expected barcode sequences. After map** to a reference genome, per-gene transcript counts were inferred based on the number of unique UMIs for each gene, after correcting for errors in the UMI sequence and excluding singleton UMIs represented by a single read only (see Methods). To assess data quality for a single-cell sequencing project, we inspected per-cell statistics as illustrated in Fig. 2b for 924 mouse Ba/F3 cells. Analyzed statistics included (1) total number of sequenced reads, (2) alignment rate, (3) number of mapped reads, (4) total number of transcripts, (5) percentage of transcripts corresponding to mitochondrial genes, and (6) number of detected genes. The percentage of unfiltered reads that could be mapped uniquely to the reference genome appeared constant across wells (~35%; Fig. 2b and Additional file 3: Figure S3a). We observed that the number of sequenced reads varied between nanowells (median 862,220; inter-quartile range 643,100–1,197,000) and increased linearly with the estimated number of captured transcripts (r = 0.99; Additional file 3: Figure S3b). This indicated that variation across nanowells was likely due to differences in cellular mRNA content or mRNA capture efficiency rather than variation in library construction or sequencing efficiency. Spatial plots of the number of detected transcripts per well did not reveal systematic effects due to well position (Additional file 4: Figure S4). Previous studies reported that mitochondrial reads may indicate poor-quality cells [19]. The percentage of mitochondrial transcripts was typically low (~6%); although we observed a higher percentage for some solid tissue samples (data not shown). We noticed that cells with poor quality data, having few detected genes and high mitochondrial content, were typically associated with low inferred transcript counts. We therefore defined a quality control criterion for processed cells by requiring a minimum number of detected transcripts for each cell. Appropriate cutoffs were determined separately for each data set (Additional file 5: Figure S5).

Sequence data from nanowell-based single-cell expression profiling. a Data analysis workflow. QC, quality control. b Quality control statistics for 924 Ba/F3 cells processed on one microchip, including total number of sequenced reads, alignment rate, number of mapped reads, total number of detected transcripts, percentage of transcripts corresponding to mitochondrial genes, and number of detected genes. Box plots indicate the interquartile range (IQR), horizontal lines are the median, whiskers extend to the most extreme data point no more than 1.5 x IQR from the box. c Median number of detected genes per cell for different sequencing depths. d Median number of detected transcripts per cell for different sequencing depths
Gene detection and reproducibility of single-cell expression profiles
To assess the sensitivity of the platform and allow comparison with other single-cell systems, we down-sampled reads for each cell and determined the median number of detected genes and transcripts with increasing sequencing depth (Fig. 2c and d). At an average sequencing depth of 100 K reads per cell we detected a median of ~2500 genes in Ba/F3 cells. The number of detected genes increased to >4000 at higher sequencing depths (Fig. 2c).
To assess the reproducibility between cells when profiling a relatively homogeneous sample type, we compared per-gene transcript counts between pairs of Ba/F3 cells. Pairwise comparisons generally showed high correlation with median r = 0.83 (Pearson correlation coefficient) and interquartile range of 0.81–0.84; an example is shown in Additional file 6: Figure S6a (r = 0.77). When performing the same analysis using per-gene read counts, we observed lower correlation (r = 0.69; Additional file 6: Figure S6b), illustrating the advantage of UMIs in reducing PCR amplification bias. Next we asked whether single-cell gene expression data accurately reflected expression profiles obtained from bulk cells. We processed total RNA from bulk Ba/F3 cells on the same microchip as Ba/F3 single cells and found that the bulk expression profile was highly correlated with the ensemble (average) of single-cell profiles (r = 0.95; Additional file 6: Figure S6c).
Assessment of cell multiplet rate and single-cell impurity
An important determinant of the utility of a single-cell profiling platform is its ability to accurately partition individual cells, such that sequencing reads for each barcode are truly derived from a single cell [Full size image
Single-cell expression profiles of cultured cell lines
We next asked if the ICELL8 system is capable of distinguishing cultured cells derived from different tissue sources. We separately dispensed eight cell suspensions of five human (A375, HCT116, NCI-H2452, Miapaca2 and KU812) and three mouse (Beta-TC6, 307 and 307-lung) cell lines across two microchips, obtaining a total of 796 human and 242 mouse cells. Principal component analyses based on the 500 most variable genes, as well as hierarchical clustering based on the 100 most variable genes, showed clear separation of different cell lines (Fig. 4a and b, Additional file 7: Figure S7). Interestingly, mouse 307 and 307-lung cells showed more intra-cluster variability likely due to the fact that these cells were derived from tumors and have undergone minimal culturing, compared to the human cell lines which have been passaged for many generations. The most variable genes included many known markers of the cell line tissues of origin. These include hemoglobin genes (HBB, HBG1, HBG2) in the peripheral blood-derived cell line KU812, MIA (melanoma inhibitory activity) in the melanoma-derived cell line A375 [21] as well as insulin (Ins1, Ins2) and islet amyloid polypeptide (Iapp) in the mouse pancreatic beta cell line Beta-TC6 (Additional file 7: Figure S7).

Single-cell RNA-seq profiling of cultured human and mouse cell lines and mouse pancreatic islets. a Unsupervised principal component analysis (PCA) for human cell lines based on the 500 most variable genes. b Unsupervised PCA for mouse cell lines based on the 500 most variable genes. c Hierarchical clustering of mouse pancreatic islet cells based on known cell type markers. Four subpopulations were identified based on four clusters indicated in the hierarchical clustering dendrogram. Cell type labels (alpha, beta, delta and PP) were assigned based on the expression of known marker genes
Identification of cell subtypes from pancreatic islets
Finally we determined whether the platform can distinguish cell types within a solid tissue sample. For this purpose we profiled 468 cells from adult mouse pancreatic islets. Pancreatic islets consist of the endocrine cells of the pancreas, with insulin-producing beta cells forming the majority. The other three major cell types include glucagon (Gcg)-producing alpha cells, somatostatin (Sst)-producing delta cells and pancreatic polypeptide (Ppy)-producing gamma cells (also known as PP cells). Hierarchical clustering based on known marker genes revealed the four distinct cell populations (Fig. 4c) and the relative abundance of cell types tracked the known composition of adult mouse pancreatic islets, including 38% beta cells (n = 179), 26% alpha cells (n = 124), 22% delta cells (n = 101), and 14% PP cells (n = 64). Unsupervised principal component analysis based on the 500 most variable genes did not show clear clusters of four major islet cell subtypes; however beta and alpha cells were largely separated (Additional file 8: Figure S8).
