
Abstract
Background
Significant gaps remain regarding the pathomechanisms underlying the autoimmune response in vitiligo (VL), where the loss of self-tolerance leads to the targeted killing of melanocytes. Specifically, there is incomplete information regarding alterations in the systemic environment that are relevant to the disease state.
Methods
We undertook a genome-wide profiling approach to examine gene expression in the peripheral blood of VL patients and healthy controls in the context of our previously published VL-skin gene expression profile. We used several in silico bioinformatics-based analyses to provide new insights into disease mechanisms and suggest novel targets for future therapy.
Results
Unsupervised clustering methods of the VL-blood dataset demonstrate a “disease-state”-specific set of co-expressed genes. Ontology enrichment analysis of 99 differentially expressed genes (DEGs) uncovers a down-regulated immune/inflammatory response, B-Cell antigen receptor (BCR) pathways, apoptosis and catabolic processes in VL-blood. There is evidence for both type I and II interferon (IFN) playing a role in VL pathogenesis. We used interactome analysis to identify several key blood associated transcriptional factors (TFs) from within (STAT1, STAT6 and NF-kB), as well as “hidden” (CREB1, MYC, IRF4, IRF1, and TP53) from the dataset that potentially affect disease pathogenesis. The TFs overlap with our reported lesional-skin transcriptional circuitry, underscoring their potential importance to the disease. We also identify a shared VL-blood and -skin transcriptional “hot spot” that maps to chromosome 6, and includes three VL-blood dysregulated genes (PSMB8, PSMB9 and TAP1) described as potential VL-associated genetic susceptibility loci. Finally, we provide bioinformatics-based support for prioritizing dysregulated genes in VL-blood or skin as potential therapeutic targets.
Conclusions
We examined the VL-blood transcriptome in context with our (previously published) VL-skin transcriptional profile to address a major gap in knowledge regarding the systemic changes underlying skin-specific manifestation of vitiligo. Several transcriptional “hot spots” observed in both environments offer prioritized targets for identifying disease risk genes. Finally, within the transcriptional framework of VL, we identify five novel molecules (STAT1, PRKCD, PTPN6, MYC and FGFR2) that lend themselves to being targeted by drugs for future potential VL-therapy.
Similar content being viewed by others


Background
Vitiligo vulgaris (non-segmental vitiligo) is a socially debilitating depigmenting disorder with a prevalence of 0.5–1% in the world population without a pronounced gender bias [1–5]. VL is an acquired, chronic skin and hair condition characterized clinically by loss of skin pigment (melanin), which, if untreated, is typically progressive and irreversible.
The autoimmune hypothesis of VL has the widest support. Autoantibodies against membranous components of melanocytes were found to be present in patient sera [6–8], and have recently been investigated as predictors of disease progression [9]. Several autoimmune comorbidities including autoimmune thyroid disease, pernicious anemia, systemic lupus erythematosus (SLE), and Addison’s disease have also been reported in association with VL [10–14]. Moreover, 20% of patients have at least one first-degree relative with VL and other autoimmune disorders (AID) [15, 16], supporting the notion of shared genetic factors across various diseases. Similarities in pathogenetic mechanisms with alopecia areata suggest new treatment modalities [17]. However, the high degree of familial aggregation has a non-Mendelian pattern of inheritance which is indicative of VL being polygenic with the influence of a large number of factors in nature [18, 19]. Several putative susceptibility loci have been reported, [20–29] including human leukocyte antigen (HLA)-associated genes such as HLA-A2, HLA-DR4 and HLA-DR7 alleles. [20, 30, 31] Non-HLA immune regulators such as ACE, CAT, CTLA4, ESR, MBL2, NALP1, FOXP1 and IL2RA [32, 33] and genes including DDR1, XBP1, NLRP1, PTPN22, and COMT have also been associated with VL [34].
The literature around VL suggests a generalized immune dysregulation that is at least in part genetically based, resulting in humoral [35] and/or cellular (T-cell) immune responses [36–40] directed at melanocytes. Both innate and adaptive immunity appear to play a role in disease progression [20]. Several antigenic proteins coded by genes such as MLANA/MART1, PMEL (melanosome related) as well as TYR, TYRP1 and TH (tyrosine related) have been identified in vitiligo. There is evidence for a key role of cytotoxic T lymphocytes (CD8 + T cells) as well as cytokines including interferon-gamma (IFN-γ) in VL pathogenesis [40–51]. Nonetheless, despite a plethora of scientific literature, the full complement of genetic elements of susceptibility, and their role in disrupting immune (and non-immune) pathways remains to be clarified.
To advance the investigation of the genetic basis for disease, we examined differential gene expression in the peripheral blood of patients diagnosed with non-segmental VL as compared with healthy control individuals and placed this information in context of our previous gene expression analysis in VL skin. We integrated the transcriptional data with functional annotations, clinical criteria and knowledge of VL genetics in an in silico bioinformatics-based approach to develop a more comprehensive framework of disease through which novel molecules could be proposed for future targeted therapy. The in silico interactome analysis allowed us to define “over-connected” key transcriptional drivers of dysregulated pathways/processes in non-segmental vitiligo. We found a total of 12 VL-blood (6) and -skin (6) transcriptional “hot spots” offering several genes that can be prioritized targets for identifying disease risk genes in future. Finally, we carefully prioritize 5 molecular targets from VL-skin or blood (STAT1, PRKCD, PTPN6, MYC and FGFR2) that can potentially explored as VL therapies.
Results
Unbiased analysis separates samples based on “disease-state”
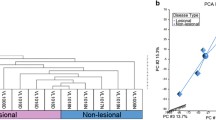
Gene expression variations from peripheral blood is sufficient to segregate VL patients from healthy controls in a non-supervised hierarchical clustering analysis (Fig. 1a). Within this “disease-state” related signature, we subsequently report a group of 319 downregulated transcripts that is significantly enriched in Gene Ontology (GO) biological processes (BPs) correlated with regulation of immune response, cell activation, response to virus, leukocyte activation as well as inflammatory response, among others (Additional file 1: Table S1). Principal components analysis (PCA) showed a spatial separation of samples without outliers or batch effects (Fig. 1b). In summation, we were able to assign a blood gene expression signature capable of separating VL patients and healthy controls using unsupervised analytical methods.

Unsupervised hierarchical clustering of VL-blood. Unsupervised hierarchical cluster analysis of the 1346 most variably expressed genes across the 14 arrays (blood samples of 8 VL patients and 6 healthy controls) was performed. a The dendrogram demonstrates a clustering of VL patients from healthy individuals revealing a molecular classification based on “disease-state”. b Principal components analysis (PCA) displays a scatter plot showing clear spatial separation of the samples (octahedrons) that are connected to a centroid sphere, with no outliers; In the 3-dimensional plot, the three principal components PC#1, #2 and #3 of all samples with all probeset IDs and their respective expression variations are plotted on the x-, y- and z- axes. The total percentage of PCA map** variability is 67.9%. Color assignations of vitiligo patient (orange) and healthy controls (green) were made after examination of distribution of samples
Pathways and processes based enrichment analyses of VL-DEGs
“Disease-state” was the largest source of variation capable of separating VL patients from healthy controls. Ninety-nine non-redundant differentially expressed genes (DEGs) (p-value <0.05, fold change (FC) cut off ≥ ±1.4) [4 up-regulated in patients (UIP) and 95 down-regulated in patients (DIP)] distinguished the two groups. The complete DEGs list is included in Additional file 2: Table S2. The top up- and down- regulated genes are included in Table 1a-b. Two members of the cytochrome P450 superfamily, CYP3A5 (FC = 1.5) and CYP1B1 (FC = 1.5), were among the top upregulated genes, and genes encoding IFN-induced proteins, IFIT1 (FC = -4.0) IF144L (FC = -4.0), IFIT3 (FC = -3.5) and IFITM2 (FC = -2.7), were among the top down-regulated genes.
DEGs were processed through an "ontology enrichment analysis" via MetaCore (Fig. 2a-d) and DAVID (Additional file 3: Table S3 a, b) to identify prominent disease-related biological pathways/processes to better understand the pathobiology underlying vitiligo (Fig. 3a-d). Canonical pathways related to immune response including IFN α and β, BCR, interleukins, antiviral actions of IFNs, and NF-κB, and stress related apoptosis were among the most enriched in the VL-blood dataset (Fig. 2a, b). The significance of male sex-related process network (Fig. 2c) is not completely understood at this time since there is no reported sex-bias for VL [1]. Enriched diseases, such as those related to connective tissue (37 DEGs), SLE (30 DEGs) and autoimmune diseases (AID) (39 DEGs) (Fig. 2d) further support the relevance of our data based on reports of comorbidity in vitiligo patients [52–54].

Functional annotation and pathway-based analysis of VL-blood associated transcriptional profile. Gene expression values from peripheral blood samples of 8 VL patients and 6 healthy individuals were compared, generating a trimmed list of 99 DEGs (4 up-and 91 down-regulated). Algorithms use the uploaded VL-blood associated DEGs as the input list and map each gene to a network object in the MetaCore database. Pathway and network analysis of the VL-blood transcriptional profile demonstrate an enrichment of the following categories: (a) canonical pathways (b) GO processes (c) process networks and (d) disease by biomarkers. Top ten enrichments are sorted and ranked by p-value shown on a logarithmic scale. A lower p-value indicates higher statistical significance

Disease-related GO pathways and processes enriched in VL-blood profile. Robust signals of: (a) immune system (51 DEGs), (b) IFN signaling and inflammation (14 DEGs), (c) response to cytokine (28 DEGs), and (d) protein catabolic process (34 DEGs), are revealed in the VL-blood DEGs. In the heat maps, red indicates upregulation while blue indicates down-regulation and white indicates unchanged expression. The blood samples cluster into VL patients (orange) and healthy controls (green). Expression value intensities are illustrated by the color of the scale with a range of -3.0 to +3.0 on a log scale
More than 50% of the VL-blood DEGs (51 DEGs; 50 DIP and 1 UIP) were associated with a prominent dysregulated immune system (Fig. 3a) which could be broadly divided into several categories: a) Type I IFN signaling (IFN α/β) [IFIT1, IFITM2, IRF9, ISG15, ISG20, MX1 MX2, OASL, PTPN6, PSMB8, (Additional file 4: Figure S1)]; b) Type II IFN (IFN-γ) signaling pathway [IRF9, PRKCD, EIF2AK2 (PKR), STAT1 (Fig. 4)]; c) IFN signaling related to inflammation [MICB, IRF9, ISG20, STAT1, TAP1, MX2, IFI44, PTPN6 (Fig. 3b)]; d) Cytokine related pathways (28 in total) (Fig. 3c) (IL-13 signaling (STAT1, PTPN6, STAT6, PRKCD] and IL-2 activation (NFKB2, HMGA1, PTPN6 and SYK, among others). Expression of the 99 DEGs was tightly coordinated across all samples supporting their use as potential biomarkers of the “disease-state” in peripheral blood of patients (Additional file 5: Figure S2).

IFN-gamma signaling pathway. The IFN-γ signaling pathway is one of the top canonical pathways enriched (p-value 0.0001, FDR 0.005) in the VL-blood profile. Histograms of down-regulated genes from VL-blood (circled in black) are demonstrated in the map. The height of the histograms corresponds to the relative expression value for a particular gene/protein. Objects and interactions as well as direction of regulation are described in the legend
Several down-regulated genes in VL patients were involved in catabolic processes such as negative regulation of cellular protein, interferon stimulated gene-15 (ISG-15) protein conjugation, and proteolysis (Fig. 3d). Additionally, FCGR2B, NCF1, FCGR1A, PRKCD, and SYK form part of the Fc gamma R-mediated phagocytosis pathway, (Additional file 3: Table S3a) and MICB, NFKB3, PSMB9, STAT1 and TAP1 are associated with antigen presentation (Fig. 2c), which are all indicative of disrupted protein breakdown accompanied by faulty antigen processing and presentation.
In silico analyses to investigate functionality of DEGs
VL-specific gene regulation was evaluated through an enrichment by “protein function” revealing a significantly high number (14) of transcription factors (TFs) and enzymes (18) (Additional file 6: Table S4). We prioritized potentially relevant individual genes, or hubs where essential genes are highly connected [55]. The topology uncovered a higher level of in-coming and out-going connections to and from the VL-dataset to the metabase with a concomitant higher clustering coefficient than either only among objects in the metabase or within the VL-blood experimental dataset (Additional file 7: Table S5a). Our analysis revealed 16 DEGs (IRF9, STAT1, FCGR2B, PRKCD, PSMB9, PSMB8, PTPN6, MX1, MX2, IFIT1, TOMM34, IFIT3, SERPING1, NCF4, TAP1 and ISG15) in the VL-blood profile (representing TFs, receptors, kinases, proteases, phosphatases, enzymes and other general proteins) that are significantly “over-connected” with objects both within the VL-blood profile as well as the larger metabase (Table 2). STAT1 and IRF9 are the most significant TFs from the VL-blood experimental dataset (Additional file 7: Table S5b) that form a central hub of in-coming and out-going (positive and negative) interactions with the “over-connected” objects (Additional file 8: Figure S3). The network includes portions of several canonical pathways such as IFN-α-IRF9-IFN-β, IFN-β-STAT1-MxA, IFN-γ-IFN-γ receptor-MxA, major histocompatibility complex (MHC class I) interactions as well as CASP8-cyt c-CASP9 tethering biological processes such as responses to immunity, defense, stress, signal transduction, apoptosis and regulation of proteolysis.
We investigated networks associated with TF encoding genes in the active dataset of potential importance to VL-blood pathology, and subjected the DEGs list to the “transcriptional regulation network” algorithm in MetaCore. Additional file 9: Table S6 summarizes 15 top scoring key TFs linked to networks tethering disease-related biological processes. Apart from significantly “over-connected” TF encoding genes such as STAT1, IRF9 and NF-kB from within the VL-blood dataset, additional but “hidden” TFs such as CREB1 MYC, IRF4, IRF1, SP1, and TP53 among others were revealed, echoing similar results obtained in our VL-skin analysis [56] where we had described MYC as a central TF in VL-pathology, as well as highlighted the relevance of significant “hidden” TFs. In summation, integrating our TF data from both VL-blood and skin (systemic and target tissue milieu) we find shared TF genes such as MYC, CREB1, TP53, SP1, ESR1, and STAT1, among others (which are either included/hidden in/from the VL-blood/skin dataset) that might be further evidence for their essential role in the transcriptional configuration underlying pathogenesis in VL. Figure 5 reveals TFs such as CREB1, MYC, IRF4, STAT1 and NF-kB as hubs of in- and out- going interactions with several down-regulated genes in the VL-blood transcriptional profile.

Network analysis of key transcriptional factors (TFs) in VL-blood pathology. The VL-blood DEGs list was analyzed using the “transcriptional regulation network” algorithm (see Additional file 9: Table S6). Auto-expanded (by one step) network was generated using the top three (CREB1, MYC and IRF4) TFs associated with disease-related BPs and then merged. STAT1 and NF-kB (from the dataset- orange boxes) as well as CREB1, MYC, IRF4, (“hidden” from dataset- yellow boxes) are TF hubs revealing a large number of curated interactions directly to and from (direction of arrow) objects in the VL-blood profile. Each hub is color coded (red = IRF4, blue = MYC, green = CREB1, magenta = STAT1/IRF9/ISG15 complex, variable colors = NF-kB (connected to several hubs). See Fig. 4 legend for types of objects
Searching for essential molecules for targeted VL-therapy
In a search for essential VL-related molecules, we selected 5 DEGs: STAT1, PRKCD and PTPN6 from the blood as well as MYC and FGFR2 from the skin transcriptional profiles. These molecules are strongly implicated as key players in the disease based upon the observations of: 1) dysregulated expression in VL skin or blood; 2) inclusion in disease-related pathways and processes; and 3) in-silico bioinformatics-based analyses highlighting functional significance. Network analysis starting with these five molecules (STAT1, PRKCD, PTPN6, MYC and FGFR2) as seed nodes, revealed them as reaction hubs with in-coming and out-going links (positive and negative influence) to several objects from the VL-skin and blood datasets as well as the larger metabase including portions of several canonical pathways (Fig. 6a). These pathways are associated with molecules such as IFN-γ, interleukins, growth and death receptors, caspases, kinases and transcriptional factors (Fig. 6b). Some of the VL-related GO biological processes tethered by these molecules are positive regulation of cellular metabolic process, response to stress, immune response, signal transduction, defense, cell proliferation as well as apoptosis, among others. Finally, the five key functional molecules are involved in a significant number of pairings with other “over-connected” objects. Additional file 10: Table S7 lists pairs of significantly connected nodes/objects within the VL-blood dataset that could interact with each other to regulate larger networks. STAT1, with 18 interactions was found to be involved in the most number of pairings, followed by PRKCD, IRF9 and ISG15.

Network analysis using key molecules in VL pathogenesis. We prioritized five DEGs (STAT1, MYC, PRKCD, PTPN6, and FGFR2) based on combined transcriptional- (VL-blood and skin) as well as in silico bioinformatics-based analyses. These 5 molecules/proteins were used as seed nodes to construct a network using the shortest path algorithm with a 2-step connection. Proteins are ordered according to their cellular localization from left to right. All five prioritized molecules are reaction hubs (blue ovals) where curated interactions are observed with both positive (green lines) and negative effects (red lines) directly to and from (direction of arrow) the hubs with several objects in the VL-blood profile as well as the larger human proteome database. (a) Portions of several canonical pathways are included in the network (in cyan), (b) a few canonical pathway portions have been traced including PTPN6-MYC-ELK1, Collagen 1-alpha11/beta1integrin-MYC, PTPN6-JAK2-BCL2 (in magenta) that are involved in immune regulation and inflammation and others such as CASP8-Cytochrome c-CASP9, Apo2L-DR5-BAX (in orange), are associated with apoptosis and survival. Gene expression data from both VL-blood and skin profiles are overlaid on the same network. The up- and down-stream interactions revealed by our network analyses may be leveraged to assess and validate functional consequences of targeting the 5 molecules by therapeutic drugs in future investigations. See Fig. 4 for legend
Chromosomal “hot spots”: comparison with literature
Next, we leveraged our VL-blood associated transcriptional profile to map chromosomal areas with an over-representation of probe expression changes as a function of presence of the disease. There is a high expectation of finding disease-associated genetic variations in such transcriptionally active regions, called “hot spots” as demonstrated earlier [56–58]. We identified 6 such regions on chromosomes 1, 6, 9, 10, 17 and 21 harboring a total of 35 down-regulated VL-blood DEGs (Fig. 7a, b). Three “hot spot’ associated genes, PSMB8 (chr6p21.3), PSMB9 (chr6p21.3) and TAP1 (chr6p21.3) overlap with putative VL susceptibility loci [56, 59, 60]. Although HLA-associated genes were not found in our transcriptional dataset, the one transcriptional “hot spot” that is overlap** in both VL-blood and -skin expression profiles, maps to chromosome 6 (chr6p24-q15) and spans the HLA region including, discoidin domain receptor tyrosine kinase 1 (DDR1; chr6p21.33) that has been reported as the strongest VL-susceptibility locus [60].

Genome-wide chromosomal distribution of VL-blood and skin DEGs. (a) Chromosomal locations of the 99 VL-blood DEGs are colored in bold black vertical bars versus the other genes which are grey. The upright bars above and below the parallel lines represent genes either on the forward or reverse strand. Each horizontal black line corresponds to one chromosome. The VL-blood DEGs were mapped to chromosomes with significant stretches, considered transcriptional “hot spots” marked by 6 red boxes. (b) A total of 35 dysregulated VL-blood genes are located in the 6 VL-blood “hot spots” with PSMB8, PSMB9 and TAP1 (bold) on chromosome 6 reported previously as potential VL-associated genetic loci. Subsequently we overlaid the 6 transcriptional “hot spots” from our previous VL-skin analysis (6 blue boxes) on the VL-blood chromosomal map. The chromosome # and locus of each transcriptional “hot spot” from VL blood (red) and skin (blue) is defined. The transcriptionally active “hot spots” on chromosome 6 overlaps between the VL-skin and blood gene expression profiles (yellow). Three additional “hot spots” from the two tissue environments map to the same chromosome but do not overlap with each other (grey)
Interestingly, the 35 DEGs map** within the 6 “hot spots” tether many of the statistically enriched immune/inflammatory canonical pathways and processes such as B-cells-, CD4+ T-cells, TNF-R2, IL-2, IL-33 and IL-17 signaling- as well as stress induced apoptosis (Additional file 11: Figure S4 a-d). The canonical IL-33 signaling pathway enriched in the 35 DEGs included in the VL-blood “hot spots” is coincident with our earlier findings in VL-skin transcriptional profile and linked to inflammation [56]. The IL-17 signaling pathway has also been increasingly implicated in several AIDs including vitiligo [61]. The remaining 64 VL-skin DEGs identified in our present analysis that did not map within a “hot spot” were enriched in immune related pathways and processes including the IFN α, β and γ, viral process, IL-4, IL-5, and IL-13 mediated inflammatory signaling, as well as negative regulation of cell proliferation. Networks starting with the 64 DEGs outside the “hotspots” tether BPs such as chromatin assembly or disassembly, collagen catabolic processes (data not shown).
Finally, we compared the full set of 99 VL-blood DEGs (35 map** inside “hot spots”, and 64 outside) to a comprehensive set of 137 VL-associated DEGs and potential gene markers compiled from several GWAS and gene expression studies. Apart from the 3 DEGs (within the “hot spots”) described above that coincided with VL-associated susceptibility genes, P2RY14 (purinergic receptor P2Y, G-protein coupled, 14) was DIP in the present dataset while being up-regulated in a recent VL-blood microarray study using pooled case and control samples [62]. No other overlaps were found with any other study.
Discussion
Major gaps remain in our knowledge regarding how genetic and environmental elements alter gene expression at the systemic level to promote or prevent vitiligo. We examined transcriptional profiles from peripheral blood of non-segmental VL cases and healthy controls (including 11 females and 3 males between the ages of 31–70 years, Table 3) using several in silico “function based” analyses to better understand underlying regulatory mechanisms in VL-pathogenesis. Our strategy was to integrate genetic, biological and clinical information to identify and characterize molecular elements of potential disease relevance in the blood. The use of unfractionated skin and PBMCs for VL-gene expression analysis offers a global and comprehensive overview of disease-associated transcriptional changes that may be relevant to disease expression and the identification of disease biomarkers.
VL-pathogenesis, similar to other heterogeneous autoimmune diseases is multifactorial and polygenic. Unsupervised clustering methods demonstrated a specific “disease-state” based molecular signature in peripheral blood distinguishing VL patients and healthy controls. This signature could potentially be leveraged to identify biomarkers for diagnostic, prognostic and classification purposes. Biological processes (including mostly down-regulated genes) such as immune/defense response and cell activation were found to be enriched in both a subset of VL-blood genes examined from the unbiased hierarchical cluster as well as the differentially expressed gene profile, reinforcing their likely importance as central drivers in disease pathogenesis. Functional annotation of the DEGs using two different analytical platforms (DAVID and MetaCore) illuminated systemically perturbed immune/defense response, protein catabolic processes and antigen processing and presentation enriched in the predominantly down-regulated genes of peripheral blood in VL patients, echoing a recent GWAS study demonstrating an enrichment of similar immuneregulation-related pathways and processes in the 48 confirmed vitiligo associated loci [29].
Broadly, our findings strongly support a down-regulation of both innate and adaptive immune/inflammatory response in VL-blood in the context of an accumulation of regulators in the activation of cytoskeletal remodeling, oxidative stress and apoptosis in VL-skin where the disease is manifest. These findings indicate tissue environment-specific changes in the disease and suggests tightly regulated mechanisms underlying disease-related pathways. The down-regulation of several immune system related pathways in VL blood might represent the susceptibility to the disease in VL patients at the systemic level. Our experiments do not address the temporal order of gene expression alterations, but rather a slice in time. Thus our VL-blood results might be expected to reveal a pattern of gene dysregulation that encompasses counter-regulatory molecules/pathways in response to an aberrant immune response.
The overlap of several DEGs within VL-blood profile and disease markers in SLE as well as other AIDs supports existing literature regarding comorbidities in vitiligo patients [10, 12, 52, 54]. Nath et al., (2001) reported evidence of a possible shared genetic effect between vitiligo and SLE, with a probable common genetic determinant on chromosome 17p13 (susceptibility gene SLEV1) [53]. In the present analysis of VL-blood, 4 dysregulated genes mapped to the “hot spot” on chromosome 17 in the neighborhood of SLEV1, indicating perturbation in the same region.
Both innate and adaptive immune response are induced by pleiotropic cytokines known as IFNs that exhibit antiviral, anti-proliferative and immunomodulatory effects. The migration of transcriptional activator STAT1 into the nucleus to activate genes involved in cell proliferation and viability can be activated by Type 1 or Type II IFN pathways [63, 64]. Previous investigations have demonstrated that IFN-γ concentrations as well as the IFN-γ: IL-10 ratio in serum plays a major role in VL-induced depigmentation and pathogenesis [47, 65]. Our analysis confirms previous reports supporting [43, 65, 66] the central role for both IFN types in vitiligo. Overall, the present study reinforces the critical role of IFN-γ-chemokine axis for the progression and maintenance of VL, and illuminates a therapeutic potential [43, 67, 68]. Furthermore, the down-regulation of TFs such as STAT1, STAT6 and NF-kB in the VL-blood dataset might be additional indication of the attempt to counter activate apoptotic signals previously observed in lesional skin. Our findings allow us to speculate about the attempt within both the systemic and the target tissue environment to repair and repopulate cells that are defective, dying or dead due to disease.
With regards to “hidden” transcriptional factors, we found several such as CREB1, MYC, IRF4, IRF1, TP53 in the VL-blood profile that echoed a similar TF circuitry found in our previous investigation of VL-lesional skin pathology [56], underscoring the importance of the mechanisms relying on the STAT1/IRF9/MYC signaling in the disease. Several transcription factors, including CREB1 and STAT3 are able to modulate the expression and/or transcriptional activity of the microphthalmia transcription factor (MITF), a master gene for melanocyte survival [69]. Our VL-skin data previously revealed a down-regulation of genes involved in terminal differentiation of melanocytes. This could be due to the lack of melanocytes in lesional skin as reported in literature [70, 71], or suggest a key role for the breakdown in the signaling pathway involving MITF and CREB1 in melanogenesis. Our results allow us to further consider the participation of the TFs such as CREB1, MYC, IRF4, STAT1/IRF9, IRF1 and TP53 in the regulation of viral processes, IFN signaling pathway, innate immune response, immune effector processes, response to cytokine stimulus, defense response, and cellular response to Type I and II IFNs. We had previously noted In VL-skin that melanocytes, immune cells and keratinocytes, among other cell types experience proliferative signals accompanied by a pronounced up-regulation of nucleotide and protein metabolic processes.
The enriched BCR pathway (including DEGs: FCGR2B, NFKB2, MAP3K7, SYK and PTPN6) that is part of the ‘adaptive’ cellular response [72] plays a critical role in the development, survival, and activation of B lymphocytes. This pathway is composed of membrane immunoglobulin molecules which bind antigens causing receptor aggregations and act through several transduction molecules such as SYK and MAP3K7 that influence TFs such as NF-kB in the nucleus, permitting several distinct outcomes, including proliferation, differentiation, apoptosis, survival and tolerance of B cells. The down-regulation of all VL-blood genes that are represented in the pathway might contribute to the switchboard functioning of the BCR pathway, enabling it to selectively turn on a specific signaling pathways by kee** others quiescent [73, 74].
Studies have demonstrated the usefulness of using gene expression data to successfully prioritize candidate genes for disease-associated single nucleotide polymorphisms in genome-wide studies [75]. Our analysis mapped 6 “hot spots” as transcriptionally active sites with TAP1 (transporter 1, ATP-binding cassette, subfamily B member), PSMB8 and PSMB9 (proteasome subunit, beta type, 8 and 9) coinciding with previously reported VL susceptibility loci [76]. These three genes are linked to macromolecular catabolic processes associated with proteasomes which normally protect against the development of T-cell mediated AIDs [77]. Proteasomes are responsible for degrading short-lived cytoplasmic proteins into peptides [78]. Among its 28 subunits, the 20S proteasome includes two subunits known as PSMB8 (LMP7) and PSMB9 (LMP2). Anomalies in TAP1 and PSMB proteins have been reported to be associated with vitiligo along with several other AIDs such as Sjogren’s syndrome, type 1 diabetes, juvenile rheumatoid arthritis, celiac disease, and multiple sclerosis [79, 80]. TAP1 functions by providing candidate peptides to MHC-I molecules within the peptide-loading complex and by transferring antigenic peptides from the cytoplasm into the endoplasmic reticulum [81]. The significance of all three proteasome-related molecules being down-regulated in blood, might indicate that coordinated events of healthy antigen processing and presentation have gone awry in VL patients. Interruption of the self-antigen presentation pathway on MHC-I has been shown to be a possible pathway for self-tolerance and autoreactivity against a variety of target organs [82, 83]. Similar to our published results in VL-skin, the “hot spot” region map** to chromosome 6 in VL-blood also covers the same HLA region coinciding with the strongest genetic associations of VL [59, 60]. The influence of the HLA region is also likely connected to its well-established role in antigen presentation and T cell activation [84].
Working towards personalized alternate therapy choices in VL, we broadened our search for key molecules related to the disease that can potentially be targeted by drugs. We focused on five DEGs (STAT1, PRKCD, PTPN6, MYC and FGFR2) based on the following criteria: 1) differential expression in VL-skin or blood; 2) inclusion in disease-related pathways; 3) interactome analysis highlighting “over-connectivity” in functional interactions; 4) network analysis demonstrating all 5 targets as interactive hubs, and 5) paired functional interacting units. Crucial regulatory roles are indicated for the two transcriptional factors (STAT1 and MYC), the phosphatase (PTPN6) and kinase (PRKCD) in VL pathogenesis. Although FGFR2 is not one of the “over-connected” DEGs, the localization of the disease-related growth receptor in the membrane with an extracellular domain capable of being recognized by specific ligands and drugs, makes it an attractive [85] molecule to investigate as a potential target. Interestingly, among the five molecules delineated by the present study (by a combination of transcriptome and bioinformatics analyses), STAT1 is presently being investigated as a promising target in the treatment of VL [68, 86]. The lack of FGFR2 expression results in a melanocyte-related disease such as piebaldism. Overall, we suggest the following five molecules to be prioritized as targets for future potential VL-therapy:
-
1)
Signal Transducer and Activator of Transcription 1 , (STAT1) (FC = -2.6; VL-blood) is a 91kDa member of the STAT family that reacts to cytokines and growth factors. It is phosphorylated by the receptor associated kinases and translocates to the cell nucleus where it acts as a transcription activator involved in apoptosis. While responding to IFN-γ, STAT1 is phosphorylated, and regulated by activation of PRKCD downstream of the activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. This can result in either pro- or anti-apoptotic outcomes depending on the cellular context and other interacting pathways [87, 88]. STAT1 has also been shown to act as a driver in cancers, modulating downstream MYC expression, which in turn promotes the capacity for proliferation, migration, and invasion of cells [89]. STAT1 activation that is essential for IFN-γ signaling has been targeted by simvastatin in mouse models (FDA-approved medication for lowering cholesterol levels) and is currently being investigated [68].
-
2)
Protein Kinase C, delta (PRKCD) (FC = -1.7; VL-blood) is a member of the family of serine- and threonine-specific protein kinases that can be activated by calcium. Human and mouse studies demonstrate that this kinase is involved in B cell signaling, regulation of growth, apoptosis, and differentiation of a variety of cell types [90], as well as the IFN-γ signaling described above [88].
-
3)
Protein Tyrosine Phosphatase, Non-Receptor Type 6 (PTPN6) (FC = -1.9; VL-blood) encodes for a member of the protein tyrosine phosphatase (PTP) family of signaling molecules that regulate a range of cellular processes such as cell growth, differentiation and mitotic cycle, while itself being regulated by the cytokine IFN-α. PTPN6 was shown to be a negative regulator of EMT transition which is involved in loss of cell-cell adhesion and increase in cell motility and reorganization in skin [91]. Cytokine-based therapies have the potential to provide novel treatments for cancer, infectious disease, and AIDS.
-
4)
We have described v-myc avian myelocytomatosis viral oncogene homolog (c-Myc/MYC) (FC = 1.5; VL-skin) as an “over-connected” TF in our previous VL-skin report [56]. It is involved in cell growth, apoptosis and metabolism. It functions as a transcription factor regulating specific target genes. The MYC proto-oncogene has been found to be stimulated in various animal and human tumors. It is important in both development as well as cell proliferation.
-
5)
Fibroblast growth factor receptor 2 (FGFR2) (FC = 1.9 VL-skin) is a highly conserved protein. It is part of the FGFR family whose members differ from each other in ligand affinities and tissue distribution. The maturation and continued existence of migrating melanoblasts is closely associated with simultaneous FGFR2 expression and a lack or dysfunction of the receptor results in maladies such as piebaldism (related to melanocyte development), among others [85]. A loss of FGFR2 function is also shown to contribute to melanoma [92]. FGFR2 has an extracellular portion that interacts with fibroblast growth factors, setting in motion a cascade of downstream signals ultimately influencing mitogenesis and differentiation.
Conclusions and future directions
The present study represents a genome-wide transcriptome analysis of blood, from predominantly female non-segmental vitiligo patients and healthy controls, examined in the context of our previous VL-skin report. Using several in silico bioinformatics-based analyses, we identify five novel molecules (STAT1, PRKCD, PTPN6, MYC and FGFR2) that have the potential to be targeted by drugs for future therapy. Additionally, we reveal molecular regulators affecting apoptosis, cytoskeletal remodeling, oxidative stress and metabolism in the skin and immune response in the blood that are suggested to contribute to the autoimmune reaction against melanocytes in the skin, similar to a recent report [29]. Future work will involve a larger cohort of different sub-phenotypes of VL patients, both male and female, accompanied with cell sorting of purified cell populations with the aim of assigning DEGs to specific candidate cell types. Longitudinal analyses of VL lesions that are newly develo**, flaring or undergoing re-pigmentation may help to illuminate specific transcriptional changes within a temporal framework of disease development and progression. On-going integrated analyses of transcriptional regulation in tissue-specific and circulatory environments has the potential to clarify details of molecular interplay that tethers the autoimmune response underlying disease pathogenesis in vitiligo. Future research into available drugs that can target the five vital disease-linked molecules proposed in this report holds the promise of expanding efficacious treatment in vitiligo.
Methods
A diagnosis of non-segmental VL (referred to as vitiligo or VL in this paper) based on established clinical criteria was the basis for recruitment to the study at the Dermatology outpatient clinic of New York Presbyterian Hospital-Cornell University. Ethical guidelines were followed by obtaining signed consent forms for punch biopsies and blood draw from patients and healthy controls (IRB # 0998-398). PBMCs were isolated from blood of VL patients and healthy control individuals that were all off therapy at the time of sampling. All specimens were snap frozen in liquid nitrogen immediately. The procedures for blood and tissue handling, PBMC extractions, total RNA preparation, cDNA synthesis have been described before [93–95]. Demographic details with age, duration of disease, ethnicity and treatment history for each subject that were chosen for subsequent analysis are presented in Table 3.
Microarray method
The Affymetrix GeneChip Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA, http://www.affymetrix.com) was followed for experimental procedures for microarray assays as described earlier [56, 94, 95]. Labeled cRNA was hybridized for 16 h at 45°C to microarrays (Affymetrix HG-U95Av1_v2). The chips were then washed, stained and scanned according to manufacturer’s protocol (Affymetrix Inc., Santa Clara, CA) on the Affymetrix Fluidics Station 750, and scanned by the Affymetrix GeneChip Scanner 3000.
Microarray data analysis
Gene expression values from lesional/non-lesional skin (n-16) and blood samples (n = 24) from VL patients and healthy controls were imported into Partek Genomics Suite v6.6 (Partek, St Louis, MO) as CEL files. Raw data preprocessing details have been described earlier [56]. The data was examined using quality control criteria in Partek software. Ten normal blood samples were discarded from the final differential expression analysis due to a failure to pass quality control check. Expression data from peripheral blood of VL patients (n = 8, 5 females and 3 males) and healthy controls (n = 6, all females) were finally used for establishing DEGs with “disease-state” as the greatest source of variation across all 12,625 probeset IDs. Average expression levels were distributed similarly across all samples.
Unsupervised hierarchical clustering
The informative probe sets with the most variation across arrays were selected for ‘unbiased’ hierarchical cluster analysis using a coefficient of variation filter greater than 0.12 across all blood arrays. We found 1346 most variably expressed probeset IDs from peripheral blood samples of VL cases and controls. A two way cluster analysis was performed on these probe sets based on Euclidean distance and centroid linkage (1-r metric) for samples and average linkage for probeset IDs. Subsequently, we examined these 1346 probeset IDs to find a set of 319 non-redundant transcripts that were down-regulated in VL patients and functionally annotated them via DAVID. Principal components analysis was performed on all 12,625 probeset IDs (transformed and normalized) to look for key variables in the dataset and observe batch effects in dataset. We superimposed sample information using color coding after assessing the sample separations by the unbiased clustering methods.
Differentially expressed genes (DEGs)
We defined DEGs between VL patients and healthy controls using similar methods as described before [56]. While controlling the p-value at <0.05, we used a FC cut off ≥ ±1.4 to generate a list of 105 DEGs. A trimmed list of 99 unique DEGs (upon removing redundancies, non-annotated probeset IDs and pseudogenes) was generated and used for all subsequent analyses. Array data is also internally validated by examining multiple probeset IDs for the same gene. When data from all such probes in the experiment are identified as correlating in an array experiment, there is higher probability of the finding being real [96]. For e.g. annotation for Probeset IDs: 263_g_at (FC-1.57), 262_at (FC = --1.50) and 36685_at (FC = -1.50) were all AMD1 (adenosylmethionine decarboxylase 1). We finally retained the representative probeset ID associated with the highest +/-fold change linked with the lowest p-value among the replicates to generate the list of non-redundant DEGs. Using both unbiased and supervised methods to reveal corroborating results serves to further bolster the disease-related findings in our report.
Tools in DAVID [97] (https://david.ncifcrf.gov/) as well MetaCore™ v6.21 (Thomson Reuters, St Joseph, MI) (http://www.genego.com) were used for ontology enrichment analyses [98] [99]. Methods were the same as described earlier [56]. We were able to generate and analyze several disease-associated canonical pathway maps in MetaCore database followed by similar investigations in the Gene Ontology (GO) and KEGG databases.
Chromosomal map** and interactome/network analyses
We leveraged the VL-blood gene expression data to delineate regions of chromosomal enrichment (“hot spots”) that contain more dysregulated genes than can be expected by chance [58, 100]. P-value ≤ 0.05 was calculated for all stretches of chromosomes that contained ≥ 3 DEGs (from the 99 VL-blood DEGs used for this analysis) and these significant stretches (“hot spots”) are outlined in boxes (Fig. 7a). The DEGs in the VL-blood expression profile were analyzed in the context of all the >12,625 genes on the array for their chromosomal location enrichment using DNA-Chip Analyzer (dChip) (www.dchip.org) using instructions as detailed in https://sites.google.com/site/dchipsoft/high-level-analysis/map-genes-to-chromosome. Duplicate probe sets were masked for gene map** using the “genome” tool as described earlier [56, 93, 95]. We subsequently overlaid the VL-skin chromosomal map on the VL-blood map to investigate overlap** “hot spots” between the two environments. In order to find key molecular elements underlying disease pathology in VL patients we used an in silico global protein interaction network analysis of the VL-blood transcriptional dataset [101]. The relative connectivity of a gene or protein reflects its functional consequence to VL [102]. It is calculated by the number of interactions between an experimental gene with the other genes on the experimental list normalized to the number of interactions it has with all genes in the human database (Metabase). One hundred and sixty VL-associated DEGs, potential biomarkers and susceptibility loci were discovered from VL-associated genes found by microarray and GWAS (http://www.genome.gov) [26, 29, 32, 33, 43, 56, 62, 70, 76, 79, 103–135] as well as the metabase and were compared to our DEGs list to search for overlaps.
Abbreviations
- AID:
-
Autoimmune disorders
- BCR:
-
B-Cell antigen receptor
- BP:
-
Biological processes
- CD8 + T:
-
Cytotoxic T lymphocytes
- DAVID:
-
Database for annotation, visualization and integrated discovery
- DEGs:
-
Differentially expressed genes
- DIP:
-
Down-regulated in patients
- FC:
-
Fold change
- GO:
-
Gene ontology
- GWAS:
-
Genome-wide association studies
- HLA:
-
Human leukocyte antigen
- IFN:
-
Interferon
- IL-2:
-
Interleukin-2
- ISG-15:
-
Interferon-stimulated gene 15
- NF-kB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- PBMC:
-
Peripheral blood mononuclear cell
- PCA:
-
Principal components analysis
- SLE:
-
Systemic lupus erythematosus
- TF/s:
-
Transcriptional factor/s
- UIP:
-
Upregulated in patients
- VL:
-
Vitiligo
References
Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ. (Editors) Fitzpatrick's Dermatology in General Medicine, vol. 1. 7th ed. New York: McGraw-Hill Companies, Inc.; 2007. p. 616–21.
Lerner AB. Vitiligo. J Invest Dermatol. 1959;32(2, Part 2):285–310.
Porter J, Beuf AH, Nordlund JJ, Lerner AB. Psychological reaction to chronic skin disorders: a study of patients with vitiligo. Gen Hosp Psychiatry. 1979;1(1):73–7.
Ongenae K, Beelaert L, van Geel N, Naeyaert JM. Psychosocial effects of vitiligo. J Eur Acad Dermatol Venereol. 2006;20(1):1–8.
Porter JR, Beuf AH, Lerner AB, Nordlund JJ. The effect of vitiligo on sexual relationships. J Am Acad Dermatol. 1990;22(2 Pt 1):221–2.
Galbraith GM, Miller D, Emerson DL. Western blot analysis of serum antibody reactivity with human melanoma cell antigens in alopecia areata and vitiligo. Clin Immunol Immunopathol. 1988;48(3):317–24.
Hertz KC, Gazze LA, Kirkpatrick CH, Katz SI. Autoimmune vitiligo: detection of antibodies to melanin-producing cells. N Engl J Med. 1977;297(12):634–7.
Naughton GK, Reggiardo D, Bystryn JC. Correlation between vitiligo antibodies and extent of depigmentation in vitiligo. J Am Acad Dermatol. 1986;15(5 Pt 1):978–81.
Jimenez-Brito G, Garza-de-La-Pena E, Perez-Romano B, Ruiz-Arguelles A. Serum Antibodies to Melanocytes in Patients With Vitiligo Are Predictors of Disease Progression. Skinmed. 2016;14(1):17–21.
Alkhateeb A, Fain PR, Thody A, Bennett DC, Spritz RA. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16(3):208–14.
Vrijman C, Kroon MW, Limpens J, Leeflang MM, Luiten RM, van der Veen JP, Wolkerstorfer A, Spuls PI. The prevalence of thyroid disease in patients with vitiligo: a systematic review. Br J Dermatol. 2012;167(6):1224–35.
Sheth VM, Guo Y, Qureshi AA. Comorbidities associated with vitiligo: a ten-year retrospective study. Dermatology. 2013;227(4):311–5.
Colucci R, Lotti F, Dragoni F, Arunachalam M, Lotti T, Benvenga S, Moretti S. High prevalence of circulating autoantibodies against thyroid hormones in vitiligo and correlation with clinical and historical parameters of patients. Br J Dermatol. 2014;171(4):786–98.
Laberge G, Mailloux CM, Gowan K, Holland P, Bennett DC, Fain PR, Spritz RA. Early disease onset and increased risk of other autoimmune diseases in familial generalized vitiligo. Pigment Cell Res. 2005;18(4):300–5.
Alikhan A, Felsten LM, Daly M, Petronic-Rosic V. Vitiligo: a comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. 2011;65(3):473–91.
Nath SK, Majumder PP, Nordlund JJ. Genetic epidemiology of vitiligo: multilocus recessivity cross-validated. Am J Hum Genet. 1994;55(5):981–90.
Rork JF, Rashighi M, Harris JE. Understanding autoimmunity of vitiligo and alopecia areata. Curr Opin Pediatr. 2016;28(4):463–69.
Ratsep R, Kingo K, Karelson M, Reimann E, Raud K, Silm H, Vasar E, Koks S. Gene expression study of IL10 family genes in vitiligo skin biopsies, peripheral blood mononuclear cells and sera. Br J Dermatol. 2008;159(6):1275–81.
Tarle RG, Nascimento LM, Mira MT, Castro CC. Vitiligo--part 1. An Bras Dermatol. 2014;89(3):461–70.
Rezaei N, Gavalas NG, Weetman AP, Kemp EH. Autoimmunity as an aetiological factor in vitiligo. J Eur Acad Dermatol Venereol. 2007;21(7):865–76.
Castanet J, Ortonne JP. Pathophysiology of vitiligo. Clin Dermatol. 1997;15(6):845–51.
Morelli JG, Norris DA. Influence of inflammatory mediators and cytokines on human melanocyte function. J Invest Dermatol. 1993;100(2 Suppl):191S–5S.
Gauthier Y, Cario Andre M, Taieb A. A critical appraisal of vitiligo etiologic theories. Is melanocyte loss a melanocytorrhagy? Pigment Cell Res. 2003;16(4):322–32.
Dell'anna ML, Picardo M. A review and a new hypothesis for non-immunological pathogenetic mechanisms in vitiligo. Pigment Cell Res. 2006;19(5):406–11.
Huang CL, Nordlund JJ, Boissy R. Vitiligo: a manifestation of apoptosis? Am J Clin Dermatol. 2002;3(5):301–8.
Spritz RA, Gowan K, Bennett DC, Fain PR. Novel vitiligo susceptibility loci on chromosomes 7 (AIS2) and 8 (AIS3), confirmation of SLEV1 on chromosome 17, and their roles in an autoimmune diathesis. Am J Hum Genet. 2004;74(1):188–91.
Chen JJ, Huang W, Gui JP, Yang S, Zhou FS, **ong QG, Wu HB, Cui Y, Gao M, Li W, et al. A novel linkage to generalized vitiligo on 4q13-q21 identified in a genomewide linkage analysis of Chinese families. Am J Hum Genet. 2005;76(6):1057–65.
Liang Y, Yang S, Zhou Y, Gui J, Ren Y, Chen J, Fan X, Sun L, **ao F, Gao M, et al. Evidence for two susceptibility loci on chromosomes 22q12 and 6p21-p22 in Chinese generalized vitiligo families. J Invest Dermatol. 2007;127(11):2552–7.
** Y, Andersen G, Yorgov D, Ferrara TM, Ben S, Brownson KM, Holland PJ, Birlea SA, Siebert J, Hartmann A, et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat Genet. 2016;48(11):1418–24.
Singh A, Sharma P, Kar HK, Sharma VK, Tembhre MK, Gupta S, Laddha NC, Dwivedi M, Begum R, Indian Genome Variation C, et al. HLA alleles and amino-acid signatures of the peptide-binding pockets of HLA molecules in vitiligo. J Invest Dermatol. 2012;132(1):124–34.
Hayashi M, ** Y, Yorgov D, Santorico SA, Hagman J, Ferrara TM, Jones KL, Cavalli G, Dinarello CA, Spritz RA. Autoimmune vitiligo is associated with gain-of-function by a transcriptional regulator that elevates expression of HLA-A*02:01 in vivo. Proc Natl Acad Sci U S A. 2016;113(5):1357–62.
Spritz RA. The genetics of generalized vitiligo and associated autoimmune diseases. Pigment Cell Res. 2007;20(4):271–8.
Spritz RA. The genetics of generalized vitiligo. Curr Dir Autoimmun. 2008;10:244–57.
Njoo MD, Westerhof W. Vitiligo. Pathogenesis and treatment. Am J Clin Dermatol. 2001;2(3):167–81.
Sandoval-Cruz M, Garcia-Carrasco M, Sanchez-Porras R, Mendoza-Pinto C, Jimenez-Hernandez M, Munguia-Realpozo P, Ruiz-Arguelles A. Immunopathogenesis of vitiligo. Autoimmun Rev. 2011;10(12):762–5.
Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16(2):90–100.
Le Poole IC, van den Wijngaard RM, Westerhof W, Das PK. Presence of T cells and macrophages in inflammatory vitiligo skin parallels melanocyte disappearance. Am J Pathol. 1996;148(4):1219–28.
van den Wijngaard R, Wankowicz-Kalinska A, Le Poole C, Tigges B, Westerhof W, Das P. Local immune response in skin of generalized vitiligo patients. Destruction of melanocytes is associated with the prominent presence of CLA+ T cells at the perilesional site. Lab Invest. 2000;80(8):1299–309.
Le Poole IC, Wankowicz-Kalinska A, van den Wijngaard RM, Nickoloff BJ, Das PK. Autoimmune aspects of depigmentation in vitiligo. J Investig Dermatol Symp Proc. 2004;9(1):68–72.
van den Boorn JG, Konijnenberg D, Dellemijn TA, van der Veen JP, Bos JD, Melief CJ, Vyth-Dreese FA, Luiten RM. Autoimmune destruction of skin melanocytes by perilesional T cells from vitiligo patients. J Invest Dermatol. 2009;129(9):2220–32.
Richmond JM, Frisoli ML, Harris JE. Innate immune mechanisms in vitiligo: danger from within. Curr Opin Immunol. 2013;25(6):676–82.
Palermo B, Campanelli R, Garbelli S, Mantovani S, Lantelme E, Brazzelli V, Ardigo M, Borroni G, Martinetti M, Badulli C, et al. Specific cytotoxic T lymphocyte responses against Melan-A/MART1, tyrosinase and gp100 in vitiligo by the use of major histocompatibility complex/peptide tetramers: the role of cellular immunity in the etiopathogenesis of vitiligo. J Invest Dermatol. 2001;117(2):326–32.
Rashighi M, Agarwal P, Richmond JM, Harris TH, Dresser K, Su MW, Zhou Y, Deng A, Hunter CA, Luster AD, et al. CXCL10 is critical for the progression and maintenance of depigmentation in a mouse model of vitiligo. Sci Transl Med. 2014;6(223):223ra223.
Wankowicz-Kalinska A, van den Wijngaard RM, Tigges BJ, Westerhof W, Ogg GS, Cerundolo V, Storkus WJ, Das PK. Immunopolarization of CD4+ and CD8+ T cells to Type-1-like is associated with melanocyte loss in human vitiligo. Lab Invest. 2003;83(5):683–95.
Badri AM, Todd PM, Garioch JJ, Gudgeon JE, Stewart DG, Goudie RB. An immunohistological study of cutaneous lymphocytes in vitiligo. J Pathol. 1993;170(2):149–55.
Tembhre MK, Parihar AS, Sharma A, Gupta S, Chattopadhyay P, Sharma VK. Participation of T cell immunoglobulin and mucin domain-3 (TIM-3) and its ligand (galectin-9) in the pathogenesis of active generalized vitiligo. Immunol Res. 2015;62(1):23–34.
Ala Y, Pasha MK, Rao RN, Komaravalli PL, Jahan P. Association of IFN-gamma : IL-10 Cytokine Ratio with Nonsegmental Vitiligo Pathogenesis. Autoimmune Dis. 2015;2015:423490.
Kemp EH, Emhemad S, Akhtar S, Watson PF, Gawkrodger DJ, Weetman AP. Autoantibodies against tyrosine hydroxylase in patients with non-segmental (generalised) vitiligo. Exp Dermatol. 2011;20(1):35–40.
Ogg GS, Rod Dunbar P, Romero P, Chen JL, Cerundolo V. High frequency of skin-homing melanocyte-specific cytotoxic T lymphocytes in autoimmune vitiligo. J Exp Med. 1998;188(6):1203–8.
van den Wijngaard R, Wankowicz-Kalinska A, Pals S, Weening J, Das P. Autoimmune melanocyte destruction in vitiligo. Lab Invest. 2001;81(8):1061–7.
Gregg RK, Nichols L, Chen Y, Lu B, Engelhard VH. Mechanisms of spatial and temporal development of autoimmune vitiligo in tyrosinase-specific TCR transgenic mice. J Immunol. 2010;184(4):1909–17.
Nejad SB, Qadim HH, Nazeman L, Fadaii R, Goldust M. Frequency of autoimmune diseases in those suffering from vitiligo in comparison with normal population. Pak J Biol Sci. 2013;16(12):570–4.
Nath SK, Kelly JA, Namjou B, Lam T, Bruner GR, Scofield RH, Aston CE, Harley JB. Evidence for a susceptibility gene, SLEV1, on chromosome 17p13 in families with vitiligo-related systemic lupus erythematosus. Am J Hum Genet. 2001;69(6):1401–6.
Zhou H, Zhao J, Tang X, Zhang X, He D. Autoimmune Hyperthyroidism, Vitiligo, Halo Nevus and Lupus. Am J Med Sci. 2016;351(2):212.
Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21(23):4205–8.
Dey-Rao R, Sinha AA. Interactome analysis of gene expression profile reveals potential novel key transcriptional regulators of skin pathology in vitiligo. Genes Immun. 2016;17(1):30–45.
Coda AB, Sinha AA. Integration of genome-wide transcriptional and genetic profiles provides insights into disease development and clinical heterogeneity in alopecia areata. Genomics. 2011;98(6):431–9.
Wu Z, Siadaty MS, Riddick G, Frierson Jr HF, Lee JK, Golden W, Knuutila S, Hampton GM, El-Rifai W, Theodorescu D. A novel method for gene expression map** of metastatic competence in human bladder cancer. Neoplasia. 2006;8(3):181–9.
** Y, Hayashi M, Fain PR, Suzuki T, Fukai K, Oiso N, Tanemura A, Holcomb CL, Rastrou M, Erlich HA, et al. Major association of vitiligo with HLA-A*02:01 in Japanese. Pigment Cell Melanoma Res. 2015;28(3):360–2.
de Castro CC S, do Nascimento LM, Walker G, Werneck RI, Nogoceke E, Mira MT. Genetic variants of the DDR1 gene are associated with vitiligo in two independent Brazilian population samples. J Invest Dermatol. 2010;130(7):1813–8.
Singh RK, Lee KM, Vujkovic-Cvi** I, Ucmak D, Farahnik B, Abrouk M, Nakamura M, Zhu TH, Bhutani T, Wei M, et al. The role of IL-17 in vitiligo: A review. Autoimmun Rev. 2016;15(4):397–404.
Wang P, Li Y, Nie H, Zhang X, Shao Q, Hou X, Xu W, Hong W, Xu A. The changes of gene expression profiling between segmental vitiligo, generalized vitiligo and healthy individual. J Dermatol Sci. 2016;84(1):40–49.
Tembhre MK, Sharma VK, Sharma A, Chattopadhyay P, Gupta S. T helper and regulatory T cell cytokine profile in active, stable and narrow band ultraviolet B treated generalized vitiligo. Clin Chim Acta. 2013;424:27–32.
Kotobuki Y, Tanemura A, Yang L, Itoi S, Wataya-Kaneda M, Murota H, Fujimoto M, Serada S, Naka T, Katayama I. Dysregulation of melanocyte function by Th17-related cytokines: significance of Th17 cell infiltration in autoimmune vitiligo vulgaris. Pigment Cell Melanoma Res. 2012;25(2):219–30.
Yang L, Wei Y, Sun Y, Shi W, Yang J, Zhu L, Li M. Interferon-gamma Inhibits Melanogenesis and Induces Apoptosis in Melanocytes: A Pivotal Role of CD8+ Cytotoxic T Lymphocytes in Vitiligo. Acta Derm Venereol. 2015;95(6):664–70.
Bertolotti A, Boniface K, Vergier B, Mossalayi D, Taieb A, Ezzedine K, Seneschal J. Type I interferon signature in the initiation of the immune response in vitiligo. Pigment Cell Melanoma Res. 2014;27(3):398–407.
Harris JE, Harris TH, Weninger W, Wherry EJ, Hunter CA, Turka LA. A mouse model of vitiligo with focused epidermal depigmentation requires IFN-gamma for autoreactive CD8(+) T-cell accumulation in the skin. J Invest Dermatol. 2012;132(7):1869–76.
Agarwal P, Rashighi M, Essien KI, Richmond JM, Randall L, Pazoki-Toroudi H, Hunter CA, Harris JE. Simvastatin prevents and reverses depigmentation in a mouse model of vitiligo. J Invest Dermatol. 2015;135(4):1080–8.
Wan P, Hu Y, He L. Regulation of melanocyte pivotal transcription factor MITF by some other transcription factors. Mol Cell Biochem. 2011;354(1-2):241–6.
Yu R, Broady R, Huang Y, Wang Y, Yu J, Gao M, Levings M, Wei S, Zhang S, Xu A, et al. Transcriptome analysis reveals markers of aberrantly activated innate immunity in vitiligo lesional and non-lesional skin. PLoS One. 2012;7(12):e51040.
Regazzetti C, Joly F, Marty C, Rivier M, Mehul B, Reiniche P, Mounier C, Rival Y, Piwnica D, Cavalie M, et al. Transcriptional Analysis of Vitiligo Skinsreveals the Alteration of WNT Pathway: A Promising Target for Repigmenting Vitiligo Patients. J Invest Dermatol. 2015;135(12):3105–14.
Dal Porto JM, Gauld SB, Merrell KT, Mills D, Pugh-Bernard AE, Cambier J. B cell antigen receptor signaling 101. Mol Immunol. 2004;41(6-7):599–613.
Geisberger R, Crameri R, Achatz G. Models of signal transduction through the B-cell antigen receptor. Immunology. 2003;110(4):401–10.
Healy JI, Goodnow CC. Positive versus negative signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1998;16:645–70.
Chen R, Morgan AA, Dudley J, Deshpande T, Li L, Kodama K, Chiang AP, Butte AJ. FitSNPs: highly differentially expressed genes are more likely to have variants associated with disease. Genome Biol. 2008;9(12):R170.
Elhawary NA, Bogari N, Jiffri EH, Rashad M, Fatani A, Tayeb M. Transporter TAP1-637G and immunoproteasome PSMB9-60H variants influence the risk of develo** vitiligo in the Saudi population. Dis Markers. 2014;2014:260732.
Zaiss DM, Bekker CP, Grone A, Lie BA, Sijts AJ. Proteasome immunosubunits protect against the development of CD8 T cell-mediated autoimmune diseases. J Immunol. 2011;187(5):2302–9.
Kincaid EZ, Che JW, York I, Escobar H, Reyes-Vargas E, Delgado JC, Welsh RM, Karow ML, Murphy AJ, Valenzuela DM, et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat Immunol. 2011;13(2):129–35.
Casp CB, She JX, McCormack WT. Genes of the LMP/TAP cluster are associated with the human autoimmune disease vitiligo. Genes Immun. 2003;4(7):492–9.
Birlea SA, ** Y, Bennett DC, Herbstman DM, Wallace MR, McCormack WT, Kemp EH, Gawkrodger DJ, Weetman AP, Picardo M, et al. Comprehensive association analysis of candidate genes for generalized vitiligo supports XBP1, FOXP3, and TSLP. J Invest Dermatol. 2011;131(2):371–81.
Parcej D, Tampe R. ABC proteins in antigen translocation and viral inhibition. Nat Chem Biol. 2010;6(8):572–80.
Faustman D, Li XP, Lin HY, Fu YE, Eisenbarth G, Avruch J, Guo J. Linkage of faulty major histocompatibility complex class I to autoimmune diabetes. Science. 1991;254(5039):1756–61.
Fu Y, Nathan DM, Li F, Li X, Faustman DL. Defective major histocompatibility complex class I expression on lymphoid cells in autoimmunity. J Clin Invest. 1993;91(5):2301–7.
** Y, Ferrara T, Gowan K, Holcomb C, Rastrou M, Erlich HA, Fain PR, Spritz RA. Next-generation DNA re-sequencing identifies common variants of TYR and HLA-A that modulate the risk of generalized vitiligo via antigen presentation. J Invest Dermatol. 2012;132(6):1730–3.
Boissy RE, Nordlund JJ. Molecular basis of congenital hypopigmentary disorders in humans: a review. Pigment Cell Res. 1997;10(1-2):12–24.
Wu CS, Komine M, Fujimoto S, Ohmatsu H, Kikuchi K, Tada Y, Yu HS, Ohtsuki M, Tamaki K. Reagents inducing epidermal proliferation also induce pigmentation: induction of keratinocyte proliferation as a novel strategy for the treatment of vitiligo. J Dermatol Sci. 2013;72(1):66–8.
Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5(5):375–86.
Deb DK, Sassano A, Lekmine F, Majchrzak B, Verma A, Kambhampati S, Uddin S, Rahman A, Fish EN, Platanias LC. Activation of protein kinase C delta by IFN-gamma. J Immunol. 2003;171(1):267–73.
Kharma B, Baba T, Matsumura N, Kang HS, Hamanishi J, Murakami R, McConechy MM, Leung S, Yamaguchi K, Hosoe Y, et al. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res. 2014;74(22):6519–30.
Mellor H, Parker PJ. The extended protein kinase C superfamily. Biochem J. 1998;332(Pt 2):281–92.
Fan LC, Shiau CW, Tai WT, Hung MH, Chu PY, Hsieh FS, Lin H, Yu HC, Chen KF. SHP-1 is a negative regulator of epithelial-mesenchymal transition in hepatocellular carcinoma. Oncogene. 2015;34(41):5252–63.
Gartside MG, Chen H, Ibrahimi OA, Byron SA, Curtis AV, Wellens CL, Bengston A, Yudt LM, Eliseenkova AV, Ma J, et al. Loss-of-function fibroblast growth factor receptor-2 mutations in melanoma. Mol Cancer Res. 2009;7(1):41–54.
Dey-Rao R, Sinha AA. Genome-wide transcriptional profiling of chronic cutaneous lupus erythematosus (CCLE) peripheral blood identifies systemic alterations relevant to the skin manifestation. Genomics. 2015;105(2):90–100.
Dey-Rao R, Sinha AA. Genome-wide transcriptional profiling data from chronic cutaneous lupus erythematosus (CCLE) peripheral blood. Data Brief. 2015;2:39–41.
Dey-Rao R, Seiffert-Sinha K, Sinha AA. Genome-wide expression analysis suggests unique disease-promoting and disease-preventing signatures in Pemphigus vulgaris. Genes Immun. 2013;14(8):487–99.
Crow MK, Wohlgemuth J. Microarray analysis of gene expression in lupus. Arthritis Res Ther. 2003;5(6):279–87.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Shmelkov E, Tang Z, Aifantis I, Statnikov A. Assessing quality and completeness of human transcriptional regulatory pathways on a genome-wide scale. Biol Direct. 2011;6:15.
Bessarabova M, Ishkin A, JeBailey L, Nikolskaya T, Nikolsky Y. Knowledge-based analysis of proteomics data. BMC Bioinf. 2012;13 Suppl 16:S13.
Luo H, Zhao X, Wan X, Huang S, Wu D. Gene microarray analysis of the lncRNA expression profile in human urothelial carcinoma of the bladder. Int J Clin Exp Med. 2014;7(5):1244–54.
Ito T, Chiba T, Yoshida M. Exploring the protein interactome using comprehensive two-hybrid projects. Trends Biotechnol. 2001;19(10 Suppl):S23–27.
Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 Suppl 1:S233–240.
Picardo M, Taieb A. In: Picardo M, Taieb A, editors. Vitiligo. Heidelberg: Springer; 2010.
Schallreuter KU, Elwary SM, Gibbons NC, Rokos H, Wood JM. Activation/deactivation of acetylcholinesterase by H2O2: more evidence for oxidative stress in vitiligo. Biochem Biophys Res Commun. 2004;315(2):502–8.
Tu CX, Gu JS, Lin XR. Increased interleukin-6 and granulocyte-macrophage colony stimulating factor levels in the sera of patients with non-segmental vitiligo. J Dermatol Sci. 2003;31(1):73–8.
Spencer JD, Gibbons NC, Bohm M, Schallreuter KU. The Ca2 + -binding capacity of epidermal furin is disrupted by H2O2-mediated oxidation in vitiligo. Endocrinology. 2008;149(4):1638–45.
Na GY, Lee KH, Kim MK, Lee SJ, Kim DW, Kim JC. Polymorphisms in the melanocortin-1 receptor (MC1R) and agouti signaling protein (ASIP) genes in Korean vitiligo patients. Pigment Cell Res. 2003;16(4):383–7.
Gottumukkala RV, Waterman EA, Herd LM, Gawkrodger DJ, Watson PF, Weetman AP, Kemp EH. Autoantibodies in vitiligo patients recognize multiple domains of the melanin-concentrating hormone receptor. J Invest Dermatol. 2003;121(4):765–70.
**a Q, Zhou WM, Liang YH, Ge HS, Liu HS, Wang JY, Gao M, Yang S, Zhang XJ. MHC haplotypic association in Chinese Han patients with vitiligo. J Eur Acad Dermatol Venereol. 2006;20(8):941–6.
Li M, Sun D, Li C, Zhang Z, Gao L, Li K, Li H, Gao T. Functional polymorphisms of the FAS gene associated with risk of vitiligo in Chinese populations: a case-control analysis. J Invest Dermatol. 2008;128(12):2820–4.
Aydingoz IE, Kanmaz-Ozer M, Gedikbasi A, Vural P, Dogru-Abbasoglu S, Uysal M. The combination of tumour necrosis factor-alpha -308A and interleukin-10 -1082G gene polymorphisms and increased serum levels of related cytokines: susceptibility to vitiligo. Clin Exp Dermatol. 2015;40(1):71–7.
Mandelcorn-Monson RL, Shear NH, Yau E, Sambhara S, Barber BH, Spaner D, DeBenedette MA. Cytotoxic T lymphocyte reactivity to gp100, MelanA/MART-1, and tyrosinase, in HLA-A2-positive vitiligo patients. J Invest Dermatol. 2003;121(3):550–6.
Abanmi A, Al Harthi F, Al Baqami R, Al Assaf S, Zouman A, Arfin M, Tariq M. Association of HLA loci alleles and antigens in Saudi patients with vitiligo. Arch Dermatol Res. 2006;298(7):347–52.
Abanmi A, Al Harthi F, Zouman A, Kudwah A, Jamal MA, Arfin M, Tariq M. Association of Interleukin-10 gene promoter polymorphisms in Saudi patients with vitiligo. Dis Markers. 2008;24(1):51–7.
Tursen U, Kaya TI, Erdal ME, Derici E, Gunduz O, Ikizoglu G. Association between catechol-O-methyltransferase polymorphism and vitiligo. Arch Dermatol Res. 2002;294(3):143–6.
Schallreuter KU, Rubsam K, Gibbons NC, Maitland DJ, Chavan B, Zothner C, Rokos H, Wood JM. Methionine sulfoxide reductases A and B are deactivated by hydrogen peroxide (H2O2) in the epidermis of patients with vitiligo. J Invest Dermatol. 2008;128(4):808–15.
Dwivedi M, Laddha NC, Shajil EM, Shah BJ, Begum R. The ACE gene I/D polymorphism is not associated with generalized vitiligo susceptibility in Gujarat population. Pigment Cell Melanoma Res. 2008;21(3):407–8.
Song GG, Bae SC, Kim JH, Lee YH. The angiotensin-converting enzyme insertion/deletion polymorphism and susceptibility to rheumatoid arthritis, vitiligo and psoriasis: A meta-analysis. J Renin Angiotensin Aldosterone Syst. 2015;16(1):195–202.
Namian AM, Shahbaz S, Salmanpoor R, Namazi MR, Dehghani F, Kamali-Sarvestani E. Association of interferon-gamma and tumor necrosis factor alpha polymorphisms with susceptibility to vitiligo in Iranian patients. Arch Dermatol Res. 2009;301(1):21–5.
Em S, Laddha NC, Chatterjee S, Gani AR, Malek RA, Shah BJ, Begum R. Association of catalase T/C exon 9 and glutathione peroxidase codon 200 polymorphisms in relation to their activities and oxidative stress with vitiligo susceptibility in Gujarat population. Pigment Cell Res. 2007;20(5):405–7.
Yasar A, Gunduz K, Onur E, Calkan M. Serum homocysteine, vitamin B12, folic acid levels and methylenetetrahydrofolate reductase (MTHFR) gene polymorphism in vitiligo. Dis Markers. 2012;33(2):85–9.
Uhm YK, Yoon SH, Kang IJ, Chung JH, Yim SV, Lee MH. Association of glutathione S-transferase gene polymorphisms (GSTM1 and GSTT1) of vitiligo in Korean population. Life Sci. 2007;81(3):223–7.
Blomhoff A, Kemp EH, Gawkrodger DJ, Weetman AP, Husebye ES, Akselsen HE, Lie BA, Undlien DE. CTLA4 polymorphisms are associated with vitiligo, in patients with concomitant autoimmune diseases. Pigment Cell Res. 2005;18(1):55–8.
Zhu Y, Wang S, Lin F, Li Q, Xu A. The therapeutic effects of EGCG on vitiligo. Fitoterapia. 2014;99C:243–51.
Mosenson JA, Eby JM, Hernandez C, Le Poole IC. A central role for inducible heat-shock protein 70 in autoimmune vitiligo. Exp Dermatol. 2013;22(9):566–9.
Dwivedi M, Laddha NC, Mansuri MS, Marfatia YS, Begum R. Association of NLRP1 genetic variants and mRNA overexpression with generalized vitiligo and disease activity in a Gujarat population. Br J Dermatol. 2013;169(5):1114–25.
Al-Shobaili HA. Update on the genetics characterization of vitiligo. Int J Health Sci. 2011;5(2):167–79.
Spritz RA. Modern vitiligo genetics sheds new light on an ancient disease. J Dermatol. 2013;40(5):310–8.
Kingo K, Aunin E, Karelson M, Philips MA, Ratsep R, Silm H, Vasar E, Soomets U, Koks S. Gene expression analysis of melanocortin system in vitiligo. J Dermatol Sci. 2007;48(2):113–22.
Li S, Yao W, Pan Q, Tang X, Zhao S, Wang W, Zhu Z, Gao J, Sheng Y, Zhou F, et al. Association analysis revealed one susceptibility locus for vitiligo with immune-related diseases in the Chinese Han population. Immunogenetics. 2015. Epub ahead of print.
Laddha NC, Dwivedi M, Mansuri MS, Singh M, Patel HH, Agarwal N, Shah AM, Begum R. Association of neuropeptide Y (NPY), interleukin-1B (IL1B) genetic variants and correlation of IL1B transcript levels with vitiligo susceptibility. PLoS One. 2014;9(9):e107020.
Spencer JD, Gibbons NC, Rokos H, Peters EM, Wood JM, Schallreuter KU. Oxidative stress via hydrogen peroxide affects proopiomelanocortin peptides directly in the epidermis of patients with vitiligo. J Invest Dermatol. 2007;127(2):411–20.
Huang Y, Yi X, Jian Z, Wei C, Li S, Cai C, Zhang P, Li K, Guo S, Liu L, et al. A single-nucleotide polymorphism of miR-196a-2 and vitiligo: an association study and functional analysis in a Han Chinese population. Pigment Cell Melanoma Res. 2013;26(3):338–47.
Manga P, Sheyn D, Yang F, Sarangarajan R, Boissy RE. A role for tyrosinase-related protein 1 in 4-tert-butylphenol-induced toxicity in melanocytes: Implications for vitiligo. Am J Pathol. 2006;169(5):1652–62.
** Y, Birlea SA, Fain PR, Ferrara TM, Ben S, Riccardi SL, Cole JB, Gowan K, Holland PJ, Bennett DC, et al. Genome-wide association analyses identify 13 new susceptibility loci for generalized vitiligo. Nat Genet. 2012;44(6):676–80.
Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–10.
Acknowledgements
We than B.K.S. and A.S. for continuous guidance and support.
Funding
This work was supported in part through grants to Dr. A.A. Sinha from the National Vitiligo Foundation and the American Vitiligo Research Foundation.
Availability of data and materials
The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus [136] and are accessible through GEO Series accession number GSE90880 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE90880).
Authors’ contributions
R.D-R: analysis of microarray data, interpretation of data, writing of manuscript, revising and final approval. A.A.S: funding, design of experiment, collection of data, critical revision and final approval.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
We have followed ethical conduct of research according to the Weill Cornell Medical College Institutional Review Board: WCM IRB # 0998-398. Singed consent forms were obtained from every patient and healthy control before obtaining punch biopsies or performing blood draws.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1: Table S1.
Potential disease relevant GO biological processes associated with down-regulated genes discovered by unsupervised hierarchical clustering. (PDF 87 kb)
Additional file 2: Table S2.
Differentially expressed genes in VL peripheral blood transcriptional profile. (PDF 320 kb)
Additional file 3: Table S3.
Ontology enrichment analysis of VL DEGs (peripheral blood). (PDF 118 kb)
Additional file 4: Figure S1.
Enriched canonical pathway related to immune response. (PDF 830 kb)
Additional file 5: Figure S2.
Profile trellis: VL-blood associated DEGs. (PDF 770 kb)
Additional file 6: Table S4.
Enrichment by Protein Function. (PDF 97 kb)
Additional file 7: Table S5.
Interactome topology and over-connected genes. (PDF 106 kb)
Additional file 8: Figure S3.
Network analysis of sixteen “over-connected” genes. (PDF 1934 kb)
Additional file 9: Table S6.
Transcriptional regulation network list from VL-blood associated experimental dataset. (PDF 196 kb)
Additional file 10: Table S7.
Significant paired nodes within over-connected functional genes. (PDF 192 kb)
Additional file 11: Figure S4.
Ontology enrichment analysis of 35 VL-blood associated DEGs that mapped to six chromosomal “hot spots”. (PDF 901 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dey-Rao, R., Sinha, A.A. Vitiligo blood transcriptomics provides new insights into disease mechanisms and identifies potential novel therapeutic targets. BMC Genomics 18, 109 (2017). https://doi.org/10.1186/s12864-017-3510-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3510-3