
Abstract
Introduction
The purpose of the present study was to systematically review the effect of cyclophosphamide treatment on pulmonary function in patients with systemic sclerosis and interstitial lung disease.
Methods
The primary outcomes were the mean change in forced vital capacity and in diffusing capacity for carbon monoxide after 12 months of therapy in patients treated with cyclophosphamide.
Results
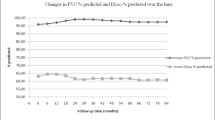
Three randomized clinical trials and six prospective observational studies were included for analysis. In the pooled analysis, the forced vital capacity and the diffusing capacity for carbon monoxide predicted values after 12 months of therapy were essentially unchanged, with mean changes of 2.83% (95% confidence interval = 0.35 to 5.31) and 4.56% (95% confidence interval = -0.21 to 9.33), respectively.
Conclusions
Cyclophosphamide treatment in patients with systemic sclerosis-related interstitial lung disease does not result in clinically significant improvement of pulmonary function.
Similar content being viewed by others

Introduction
Scleroderma (systemic sclerosis (SSc)) is an autoimmune connective tissue disorder characterized by microvascular injury, excessive fibrosis of the skin and distinctive visceral changes that can involve the lungs, heart, kidneys and gastrointestinal tract [1]. Interstitial lung disease (ILD) occurs in patients who have CREST (Calcinosis, Raynaud, ESophagitis, Telangiectases), limited cutaneous systemic sclerosis-lcSSc and diffuse cutaneous scleroderma (dcSSc), but it is somewhat more common in patients who have diffuse disease [2, 3]. The ILD that occurs in scleroderma patients includes a number of entities, as summarized in Table 1[4]. The prevalence of ILD in scleroderma varies from 25% to 90% depending on the ethnic background of the patients studied and on the method used to detect the ILD [5].
Pulmonary function tests with evaluation of the forced vital capacity (FVC), the total lung capacity and the diffusing lung capacity of carbon monoxide (DLCO), chest radiography and high-resolution computed tomography are common clinical tests used to evaluate lung disease in scleroderma. Imaging reveals fibrotic changes of lung parenchyma. Previous research has found pulmonary function tests to reveal a restrictive pattern in 23% of patients with limited disease, and found 40% of patients with diffuse disease to have pulmonary fibrosis [4, 5]. ILD as assessed by chest radiography has been reported in 33% of patients with limited scleroderma and in 40% of patients with diffuse SSc [5]. High-resolution computed tomography detects ILD changes in 90% to 100% of SSc patients [2, 5].
ILD is associated with increased mortality in patients who have SSc. The greatest loss of lung volume occurs within the first 2 years of the disease, and pulmonary-related deaths occur with greater frequency in the second 5 years from disease onset [5]. Patients with severe lung involvement (defined as FVC < 55% and DLCO < 40% of predicted) have a worse prognosis, with a mortality of 42% within 10 years of the onset of disease [5].
A number of agents have been evaluated for treatment of SSc-related ILD but none have proven effective in altering the disease course. Cyclophosphamide (CYC) is a cytotoxic immunosuppressive agent that suppresses lymphokine production and modulates lymphocyte function by alkylating various cellular constituents and depressing the inflammatory response. Of all the drugs studied for the treatment of SSc-related ILD, only CYC has shown much promise of benefit in slowing down the decrease in, or even improving, lung function and survival [1]. Retrospective studies, pilot studies, and open-label clinical trials support the effectiveness of CYC therapy in preventing a decline in lung function and premature death in patients with SSc and ILD.
Despite these individual study results, previous systematic reviews of retrospective studies of the CYC effect in SSc lung disease have yielded conflicting results, suggesting either some or no benefit of this agent [6, 7]. To determine the possible benefit of CYC as management for SSc-related ILD, we examined the benefit of CYC on lung function as measured by pulmonary function tests by conducting a systematic review and meta-analysis of randomized clinical trials and prospective observational studies in patients with SSc treated with CYC.
Materials and methods
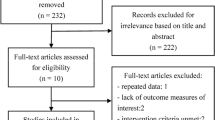
The study selection, assessment of eligibility criteria, data extraction and statistical analysis were performed based on a prespecified protocol according to the Cochrane Collaboration guidelines [8]. The present article has been prepared in accordance with the QUOROM statement [9]. An expert medical librarian searched Ovid EMBASE, Ovid MEDLINE, and the Ovid Cochrane Library from 1986 to 2008 using the terms systemic scleroderma, autoimmune diseases, cyclophosphamide, immunosuppressive therapies, interstitial lung disease, randomized controlled trials, observational studies, multicenter studies, clinical trials phase II, clinical trials phase III, and clinical trials phase IV.
To locate unpublished trials, we searched the electronic abstract databases of the annual scientific meetings of the European League Against Rheumatism, the American College of Rheumatology and the American Thoracic Society, from the approval of CYC as a treatment for autoimmune disease in 1986 to the present. No restriction for language was used.
Assessment of eligibility criteria for inclusion or exclusion and extraction of outcome variables was performed independently by two investigators (CN and ELM) with an intraobserver agreement kappa statistic of 1.
Selection and outcomes
We selected randomized clinical trials [1, 10, 11] and prospective observational studies [12–18] that included patients classified as having limited and/or diffuse SSc according to the American College of Rheumatology criteria [19] and a diagnosis of ILD [20] treated with oral or intravenous CYC. The dose of CYC administered differed across the various cohorts of patients. Some studies expressed the CYC dose in milligrams per kilogram per day and others in milligrams per square meter of body surface. The oral dose of CYC ranged from 1 mg/kg/day to 2.5 mg/kg/day, and the intravenous dose of CYC ranged from 500 mg/m2 to 750 mg/m2 – except for one study in which 900 mg/kg/day intravenous CYC was administered (Tables 2 and 3).
In the randomized clinical trials, patients were randomly allocated to receive treatment with CYC versus placebo [1, 10] or versus azathioprine [11] for at least 12 months. In the observational prospective studies, scleroderma patients were treated with CYC for at least 12 months, and were evaluated at baseline and after 12 months of therapy. Corticosteroid treatment was permitted in both the randomized clinical trials and observational studies.
A clinically important change between two groups of treatment (CYC versus non-CYC) has been previously reported as an improvement ≥ 10% of the predicted value at 12 months or from the baseline value of FVC or DLCO [12, 11]. Previous studies have reported no or very mild adverse events in patients with SSc-related ILD who were treated with CYC [26, 27]. Other studies have reported bladder complications secondary to the drug in patients with SSc [28, 29]. The adverse events counted in our nine studies included two cases of hemorrhagic cystitis [17] and several cases of hematuria – one case in Valentini and colleagues [18], two cases in Hoyles and colleagues [10] and nine cases in Tashkin and colleagues [1]; bladder cancer was not reported. A doubling of bladder cancer risk in Wegener's granulomatosis patients for every 10 g increase in the cumulative dose of CYC and an eightfold increased risk for treatment duration longer than 1 year has been reported [30]. Since the results of our meta-analysis are based on 12 months of follow-up they may not reflect adverse events develo** over longer durations of treatment or follow-up.
Our study has additional limitations. The number of patients enrolled, the dose of CYC, concomitant corticosteroid use, the SSc-related ILD disease extent and SSc disease duration, and the comparator treatments varied across studies. For example, some evidence suggests that glucocorticoids may be effective in SSc-related ILD in certain situations [5, 25, 31–33]. There may be other factors contributing to heterogeneity unidentified by our review. The shortage of randomized controlled trials on this topic is a limitation, and larger randomized controlled trials are needed to better understand the role of CYC in the care of these patients. In our meta-analysis, two of the three greatest mean differences of the FVC after 12 months of therapy were achieved in observational studies using higher doses of corticosteroids [15, 16], limiting our ability to draw a clear conclusion of beneficial effect of CYC alone. It is also possible that azathioprine has a beneficial treatment effect, which would reduce the magnitude of difference in benefit seen in comparison with CYC. A further limitation is that several studies, particularly the observational studies, had small numbers of patients.
Conclusions
Based on available data, CYC treatment in patients with SSc-related ILD does not appear to result in clinically significant improvement of pulmonary function. Since none of the patients included in these studies were selected on the basis of progression of ILD or the time from the SSc-related ILD diagnosis, further randomized clinical studies are needed to evaluate whether CYC (or any) therapy might exert a beneficial effect in patients with worsening ILD. It is possible, for example, that patients treated sooner after diagnosis or at earlier stages of SSc-related ILD might have a better response to CYC treatment. Based on current understanding, however, SSc-related ILD will be only effectively addressed when better understanding of the immunopathophysiology of the disease is understood and when treatment options more effective than CYC become available.
Abbreviations
- ACR:
-
American College of Rheumatology
- AZA:
-
azathioprine
- BAL:
-
bronchoalveolar lavage
- CI:
-
confidence interval
- CREST:
-
Calcinosis, Raynaud, ESophagitis, Sclerodactylia, Telangiectases
- CT:
-
computed tomography
- CXR:
-
chest radiography
- CYC:
-
cyclophosphamide
- dcSSC:
-
diffuse cutaneous systemic sclerosis
- DLCO:
-
diffusing lung capacity of carbon monoxide
- EULAR:
-
European League Against Rheumatism
- FVC:
-
forced vital capacity
- HRTC:
-
high resolution computed tomography
- ILD:
-
Interstitial Lung Disease
- IV:
-
intravenous
- lcSSc:
-
limited cutaneous systemic sclerosis
- PFT:
-
pulmonary function test
- RR:
-
relative risk
- SE:
-
standard error
- SSC:
-
systemic sclerosis
- SSc-ILD:
-
systemic sclerosis related interstitial lung disease
- TLC:
-
total lung capacity.
References
Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, Arriola E, Silver R, Strange C, Bolster M, Seibold JR, Riley DJ, Hsu VM, Varga J, Schraufnagel DE, Theodore A, Simms R, Wise R, Wigley F, White B, Steen V, Read C, Mayes M, Parsley E, Mubarak K, Connolly MK, Golden J, Olman M, Fessler B, Rothfield N, et al: Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. 2006, 354: 2655-2666. 10.1056/NEJMoa055120.
Van Laar JM, Stolk J, Tyndall A: Scleroderma lung. Pathogenesis, evaluation and current therapy. Drugs. 2007, 67: 985-996. 10.2165/00003495-200767070-00004.
Bérezné A, Valeyre D, Ranque B, Guillevin L, Mouthon L: Interstitial lung disease associated with systemic sclerosis. What is the evidence for efficacy of cyclophosphamide?. Ann N Y Acad Sci. 2007, 1110: 271-284. 10.1196/annals.1423.029.
Strange C, Highland KB: Interstitial lung disease in the patient who has connective tissue disease. Clin Chest Med. 2004, 25: 549-559. 10.1016/j.ccm.2004.05.009.
White B: Interstitial lung disease in scleroderma. Rheum Dis Clin North Am. 2003, 29: 371-390. 10.1016/S0889-857X(03)00025-5.
Latsi PI, Wells AU: Evaluation and management of alveolitis and interstitial lung disease in scleroderma. Curr Opin Rheumatol. 2003, 15: 748-755. 10.1097/00002281-200311000-00011.
Zandman-Goddard G, Tweezer-Zaks N, Shoenfeld Y: New therapeutic strategies for systemic sclerosis: a critical analysis of the literature. Clin Dev Immunol. 2005, 12: 165-173. 10.1080/17402520500233437.
Cochrane Website. [http://www.cochrane.org/resources/handbook/index.htm]
Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF: Improving the quality of reports of meta-analyses of randomised controlled trials: the QUOROM statement. QUOROM Group. Br J Surg. 2000, 87: 1448-1454. 10.1046/j.1365-2168.2000.01610.x.
Hoyles RK, Ellis RW, Wellsbury J, Lees B, Newlands P, Goh NS, Roberts C, Desai S, Herrick AL, McHugh NJ, Foley NM, Pearson SB, Emery P, Veale DJ, Denton CP, Wells AU, Black CM, du Bois RM: A multicenter, prospective, randomized, double blind, placebo-controlled trial of corticosteroids and intravenous cyclophosphamide followed by oral azathioprine for the treatment of pulmonary fibrosis in scleroderma. Arthritis Rheum. 2006, 54: 3962-3970. 10.1002/art.22204.
Nadashkevich O, Davis P, Fritzler M, Kovalenko W: A randomized unblinded trial of cyclophosphamide versus azathioprine in the treatment of systemic sclerosis. Clin Rheumatol. 2006, 25: 205-212. 10.1007/s10067-005-1157-y.
Davas EM, Peppas C, Maragou M, Alvanou E, Hondros D, Dantis PC: Intravenous cyclophosphamide pulse therapy for the treatment of lung disease associated with scleroderma. Clin Rheumatol. 1999, 18: 455-461. 10.1007/s100670050138.
White B, Moore WC, Wigley FM, **ao HQ, Wise RA: Cyclophosphamide is associated with pulmonary function and survival benefit in patients with scleroderma and alveolitis. Ann Intern Med. 2000, 132: 947-954.
Beretta L, Caronni M, Raimondi M, Ponti A, Viscuso T, Origgi L, Scorza R: Oral cyclophosphamide improves pulmonary function in scleroderma patients with fibrosing alveolitis: experience in one centre. Clin Rheumatol. 2007, 26: 168-172. 10.1007/s10067-006-0254-x.
Airò P, Danieli E, Rossi M, Frassi M, Cavazzana I, Scarsi M, Grottolo A, Franceschini F, Zambruni A: Intravenous cyclophosphamide for interstitial lung disease associated to systemic sclerosis: results with an 18 month long protocol including a maintenance phase. Clin Exp Rheumatol. 2007, 25: 293-296.
Pakas I, Ioannidis JP, Malagari K, Skopouli FN, Moutsopoulos HM, Vlachoyiannopoulos PG: Cyclophosphamide with low or high dose prednisolone for systemic sclerosis lung disease. J Rheumatol. 2002, 29: 298-304.
Silver RM, Warrick JH, Kinsella MB, Staudt LS, Baumann MH, Strange C: Cyclophosphamide and low-dose prednisone therapy in patients with systemic sclerosis (scleroderma) with interstitial lung disease. J Rheumatol. 1993, 20: 838-844.
Valentini G, Paone C, La Montagna G, Chiarolanza I, Menegozzo M, Colutta E, Ruocco L: Low-dose intravenous cyclophosphamide in systemic sclerosis: an open prospective efficacy study in patients with early diffuse disease. Scand J Rheumatol. 2006, 35: 35-38. 10.1080/03009740510026896.
Subcommittee for Scleroderma Criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee: Preliminary criteria for the classification of systemic sclerosis (scleroderma). Arthritis Rheum. 1980, 23: 581-590. 10.1002/art.1780230510.
American Thoracic Society, European Respiratory Society: American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Resp Crit Care Med. 2002, 165: 277-304.
Guidelines for the measurement of respiratory function: recommendations of the British Thoracic Society and the Association of Respiratory Technicians and Physiologists. Respir. 1994, 88: 165-194. 10.1016/S0954-6111(05)80346-4.
Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, McQuay HJ: Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Control Clin Trials. 1996, 17: 1-12. 10.1016/0197-2456(95)00134-4.
Wells GA, Shea B, O'Connell D: The Newcastle-Ottawa scale (NOS) for assessing the quality of nonrandomized studies in meta-analysis. Proceedings of the Third Symposium on Systematic Reviews: Behind the Basics. Oxford. England, July 4–9. 2000, [http://www.ohri.ca/programs/clinical_epidemiology/oxford.htm]
Kazerooni EA, Martinez FJ, Flint A, Jamadar DA, Gross BH, Spizarny DL, Cascade PN, Whyte RI, Lynch JP, Toews G: Thin-section CT obtained at 10-mm increments versus limited three-level thin-section CT for idiopathic pulmonary fibrosis: correlation with pathologic scoring. AJR Am J Roentgenol. 1997, 169: 977-983.
Cohen J: A power primer. Psychol Bull. 1992, 112: 155-159. 10.1037/0033-2909.112.1.155. [http://psycnet.apa.org/index.cfm?fa=search.displayRecord&uid=1992-37683-001]
Steen VD, Lanz JK, Conte C, Owens GR, Medsger TA: Therapy for severe interstitial lung disease in systemic sclerosis. A retrospective study. Arthritis Rheum. 1994, 37: 1290-1296. 10.1002/art.1780370904.
Tashkin DP, Elashoff R, Clements PJ, Roth MD, Furst DE, Silver RM, Goldin J, Arriola E, Strange C, Bolster MB, Seibold JR, Riley DJ, Hsu VM, Varga J, Schraufnagel D, Theodore A, Simms R, Wise R, Wigley F, White B, Steen V, Read C, Mayes M, Parsley E, Mubarak K, Connolly MK, Golden J, Olman M, Fessler B, Rothfield N, et al: Effect of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007, 176: 1026-1034. 10.1164/rccm.200702-326OC.
Giacomelli R, Valentini G, Salsano F, Cipriani P, Sambo P, Conforti ML, Fulminis A, De Luca A, Farina G, Candela M, Generini S, De Francisci A, Tirri E, Proietti M, Bombardieri S, Gabrielli A, Tonietti G, Cerinic MM: Cyclophosphamide pulse regimen in the treatment of alveolitis in systemic sclerosis. J Rheumatol. 2002, 29: 731-736.
Plotz PH, Klippel JH, Decker JL, Grauman D, Wolff B, Brown BC, Rutt G: Bladder complications in patients receiving cyclophosphamide for systemic lupus erythematosus and rheumatoid arthritis. Ann Intern Med. 1979, 91: 221-223.
Talar-Williams C, Hijazi YM, Walther MM, Linehan WM, Hallahan CW, Lubensky I, Kerr GS, Hoffman GS, Fauci AS, Sneller MC: Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegner granulomatosis. Ann Intern Med. 1996, 124: 477-484.
Knight A, Askling J, Granath F, Sparen P, Ekbom A: Urinary bladder cancer in Wegener's granulomatosis: risks and relation to cyclophosphamide. Ann Rheum Dis. 2004, 63: 1307-1311. 10.1136/ard.2003.019125.
Griffiths B, Miles S, Moss H, Robertson R, Veale D, Emery P: Systemic sclerosis and interstitial lung disease: a pilot study using pulse intravenous methylprednisolone and cyclophosphamide to assess the effect on high resolution computed tomography scan and lung function. J Rheumatol. 2002, 29: 2371-2378.
Pai BS, Srinivas CR, Sabitha L, Shenoi SD, Balachandran CN, Acharya S: Efficacy of dexamethasone pulse therapy in progressive systemic sclerosis. Int J Dermatol. 1995, 34: 726-728. 10.1111/j.1365-4362.1995.tb04664.x.
Acknowledgements
The authors would like to thank Dr Victor Montori and Dr Hassan Murad for their expertise and advice in the conduct of this study. There was no funding support for the present study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CN conceived the study and participated in its design, coordination, data acquisition and analysis, and in manuscript preparation. CPW and ELM participated in the study design, data acquisition and analysis, and in manuscript preparation. PJE participated in data acquisition and in manuscript preparation. All authors read and approved the final manuscript.
An erratum to this article is available at http://dx.doi.org/10.1186/ar2679.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Nannini, C., West, C.P., Erwin, P.J. et al. Effects of cyclophosphamide on pulmonary function in patients with scleroderma and interstitial lung disease: a systematic review and meta-analysis of randomized controlled trials and observational prospective cohort studies. Arthritis Res Ther 10, R124 (2008). https://doi.org/10.1186/ar2534
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/ar2534