
Abstract
Polyglutamine (polyQ) diseases are a group of neurodegenerative conditions, induced from CAG trinucleotide repeat expansion within causative gene respectively. Generation of toxic proteins, containing polyQ-expanded tract, is the key process to cause neurodegeneration. Till now, although polyQ diseases remain uncurable, numerous therapeutic strategies with great potential have been examined and have been proven to be effective against polyQ diseases, including diverse small biological molecules and many pharmacological compounds mainly through prevention on formation of aggregates and inclusions, acceleration on degradation of toxic proteins and regulation of cellular function. We review promising therapeutic strategies by using Drosophila models of polyQ diseases including HD, SCA1, SCA3 and SBMA.
Similar content being viewed by others


Introduction
Among the unstable repeat expansion disorders, the dominantly inherited polyQ diseases caused by CAG repeat expansions within responsible genes are the most common group [1]. The first discovered polyQ disorder was Spinal bulbar muscular atrophy (SBMA), also known as Kennedy’s disease which is X-linked, in 1991, a dynamic repeat mutation in the androgen receptor (AR) gene [2]. Since then at least eight additional polyQ diseases have been identified. To date, this group includes the distinct Spinocerebellar Ataxias (SCA1, SCA2, SCA3/Machado–Joseph disease, SCA6, SCA7 and SCA17) [3–5], dentatorubral-pallidoluysian atrophy (DRPLA), Huntington disease (HD), and most recently, Huntington disease-like 2 (HDL2) [6]. Generally, compared with other neurodegenerative diseases, polyQ diseases have low prevalence. 1 ~ 3/100 000 Europeans have autosomal dominant cerebellar ataxias (ADCAs) [7], such as SCA1, SCA2, SCA3/MJD, SCA6, SCA7 and SCA17, while 1.6/100 000 individuals have SBMA [8] and 5 ~ 7/100 000 white people suffered HD [9].
In all polyQ disorders, the abnormally translated polyglutamine domain in the corresponding disease protein would lead to dysfunction and pathogenic effects, when the CAG repeat expansion in respective causative genes surpasses a pathological threshold. The involvement of the polyQ-expanded domain in pathogenic mechanisms has been illustrated in various ways. For toxic effects, several intracellular molecular mechanisms have been illustrated, including formation of aggregates [10–14], dysregulation of cellular protein homeostasis [15–18], alternations in transcription [19–24], impairment of axonal transport [25, 26], mitochondrial dysfunction [27–29] and harassment of intracellular Ca2+ homeostasis [30–34]. PolyQ disorders share numerous common traits including progressive neurodegeneration in specific neuronal populations, formation of protein aggregates and the negative correlation between the number of CAG repeats and the age of onset which means the greater number of such repeats leads to the earlier onset of the diseases [35–38]. Despite these common aspects, however, there are respective aspects to polyQ diseases as well. Each polyQ disease is a distinctive disorder with characteristic symptomatic profiles and different neurodegeneration occurring in particular brain regions. Although the expanded proteins are prevalently expressed throughout the CNS, there are two remarkable exceptions: SCA6 and Kennedy’s disease. The CACNA1A calcium channel subunit in SCA6 is primarily expressed in affected cerebellar Purkinje cells, while the AR in Kennedy’s disease is principally expressed in vulnerable motor neurons. Additionally, the various properties of each of PolyQ disease proteins differ in subcellular localization, structure, activity and biological function, suggesting that the specific details of pathogenic effects may be unique to each disease [2].
Review
Advantages of drosophila as genetic models for polyQ disease
Application of animal models is a powerful approach to address some of the outstanding questions underlying polyQ disease. Although modelling neurological diseases in rodent has been of significant impact, using drosophila melanogaster model offers many advantages for pathogenic mechanism and therapeutic compounds. Due to the shorter life span, rapid reproductive cycle, accessibility of several techniques and tools to modulate gene expression, and comparatively well-known anatomy and phenotypes, fruit fly prefer faster polyQ modelling [39, 40]. In addition, using GAL4/UAS system [41], we can manipulate the expression level of transgene and expression in specific cell types [42]. Drosophila and human genomes are characterized a high degree of conservation in fundamental biological pathways [43]. With the few chromosome number, convenient genetic operation, simple inherited background, drosophila model has great value in functional analysis of human disease genes. Moreover, high-throughput pharmalogical screens are also possible due to the minimal barrier in central nervous system, although flies have a complex nervous system and brain [44]. Up till now, several Drosophila models of polyQ diseases have been developed, including SCA1, SCA2, SCA3/MJD, SCA7, SCA17, DRPLA, HD and SMBA. Moreover, several human pathological features, including formation of inclusions, neural degeneration, motor dysfunction and premature lethality, have successfully been recapitulated in Drosophila models of polyQ [45–49].
Therapeutic strategies for polyQ diseases
According to pathogenic hypothesis mentioned above, therapeutic strategies could target on the general toxic mechanism triggered by expanded polyQ. Generally, therapeutic strategies classified into targeting prevention on formation of aggregates and inclusions, targeting acceleration on degradation of toxic proteins and regulation of cellular function (Table 1).
Prevention on formation of aggregates and inclusions
Insoluble aggregates present toxic effects for neurons and result in cell death and organism pathology [11]. PolyQ diseases seem to be originated by proteolytic cleavage of mutant protein containing polyQ-expanded tract to form toxic fragments [9, 10], which aberrantly fold into amyloid-like aggregates (oligomers), then assemble into nuclear and cytoplasmic deposits which are the cellular hallmarks of polyQ diseases. The neuronal protein aggregates mainly present in nucleus (SCA1, SCA2, SCA3/MJD, SCA7, SCA17 and DRPLA), cytoplasm (SCA6) or both cytoplasm and nucleus (HD and SBMA) (Figure 1) [76, 77].

The intra-cellular sites of toxic effects of proteins in polyQ disorders. PolyQ aggregates induce cytotoxic effects through a range of mechanisms. To form insoluble inclusions, compiling of polyQ-expanded protein provoke quality-control mechanisms including ubiquitin–proteasome system, chaperones and autophagy. By interacting with transcriptional factors, the toxic polyQ proteins could regulate transcriptional processes. Other cellular sites of toxic effects induced by polyQ mutant proteins include Ca2+ channel, mitochondria, and cytoskeleton with diverse abnormalities respectively.
RNA interference-based therapeutics
Mutant polyglutamine protein expression could be inhibited by RNA interference (RNAi). Inhibiting formation of mutant protein containing expanded polyQ tract, RNAi is the most straightforward approach to selectively decrease expression of the mutant allele. **a et al. and Harper et al. have successfully testified that administration of RNAi, against ATXN1 or IT15, improved the motor impairment in SCA1 and HD transgenic mice models [78, 79]. Similarly, in several Drosophila models of human polyQ disorders including SCA3/MJD, SCA1 and HD, Mallik et al. [50] discovered that RNAi suppress pathogenesis of polyQ diseases by down-regulating transcripts of hsr-ω, a dominant modifier for polyQ pathogenic mechanism found in Drosophila model of SCA1 [42]. Co-expressing hsrω-RNAi transgenes suppresses expanded polyQ-induced eye-specific degeneration, diminishes toxicity of mutant polyQ protein in nervous system and inhibits polyQ protein aggregation.
Therapeutic pharmacological compounds
A variety of chemical compounds with great potential have been proven effective for treatment of polyQ diseases by using Drosophila model of HD through preventing polyQ-expanded aggregation. Cystamine, methylene blue (MB), camptothecin, OH-camptothecin, 18b-glycyrrhetinic acid and carbenoxolone are promising therapeutics for HD. Cystamine, a competitive inhibitor of tissue transglutaminase (tTG), was thought to reduce Huntingtin (Htt) aggregation by interfering with tTG-mediated glutamine crosslinking [51, 52]. Bortvedt el at discovered that in adult HD flies co-treatment of feeding cystamine and expressing a transgene encoding the anti-htt intracellular antibody (intrabody) C4-scFv leads to alleviation of photoreceptor neurodegeneration without benefit in longevity, however in larval and adult stages of Drosophila feeding cystamine showed opposite effect: longevity was prolonged without photoreceptor rescue [53]. MB has reported promotion of degradation androgen receptor polypeptides [80] and inhibition of mutant Htt aggregation [81]. Sontag et al. found that administrating MB to Drosophila of HD significantly increased the rhabdomeres number accompany with a decrease in Htt-mediated neurodegeneration. In addition, treating MB in the larval stages reduced the aggregates number by 87% and diminished the aggregates size, suggesting that MB could be promising therapeutic drug for HD through the regulation of aggregation [54]. Additionally, Schulte el at by screening the subcellular distribution of Htt labeled by mRFP and monitoring the morphology of cultured neurons via live-imaging discovered 18b-glycyrrhetinic acid, as well as camptothecins, OH-camptothecin, carbenoxolone inhibited formation of Htt aggregate, restored neurite morphology and viability in Drosophila HD model [55].
Molecular therapeutics
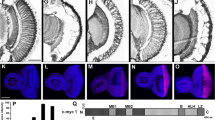
Accumulation of aggregates to form inclusions recruits cellular normal proteins, such as molecular chaperones, which suggests that polyQ-expanded domain changes protein structure and activates chaperones against protein misfolding [82]. The finding that expanded polyQ tract is capable of transiting into diverse conformations is also affirmed by evidence that, in fly (SCA3/MJD, HD) and mouse (SCA1) models, overexpression of molecular chaperones represses toxicity [83, 84]. Thus, several biological molecules may thought to be potential therapeutics targeting prevention on formation of pathogenic aggregates and toxic structure of polyQ proteins, such as polyglutamine binding peptide 1 (QBP1), a peptide P42, chaperones heat shock proteins including heat shock protein 70 (Hsp70), Hsp40, Hsp110 and Hsp22. Application of QBP1 which is capable of selectively binding to the polyQ-expanded stretch suppresses compound eye degeneration, polyQ aggregates formation and rescues premature death in SCA3/MJD Drosophila model [56], suggesting that QBP1 is a potential therapeutic molecule on polyQ disorders. P42, a 23 amino acid-long peptide which is a part of the endogenous Htt protein, plays a protective role in preventing polyQ-hHtt aggregation, improving the impaired axonal transport by restoring the total number and motion of vesicles, ameliorating behavioral dysfunctions and against polyQ-hHtt induced toxicity in HD Drosophila model. However, no protective effects were found in other polyQ diseases [57]. Although the toxic conformation of polyQ proteins remains elusive, therapeutic strategy targeting on toxic structure of expanded polyglutamine proteins may be a promising approach against untreatable polyQ disease. In Drosophila melanogaster model of SCA3/MJD, co-expressing the human gene HSPA1L which encodes Hsp70, Warrick et al. discovered that Hsp70 completely rescued external eye pigmentation, partially restored retinal structure of brain, and partially restored adult viability, suggesting that molecular chaperone Hsp70 suppresses polyglutamine-induced neurodegeneration and toxicity as well as indicating that HSP70 would be a promising candidate with great potential as a treatment method [58]. Additionally, in a Drosophila model of SBMA, Adrienne et al. demonstrated that Hsp70 with its co-chaperone Hip which enhances Hsp70 binding to its substrates accelerated polyQ AR clearance, and identified that Hsp70 with YM-1, a synthetic co-chaperone that acts similarly to Hip, also enhanced polyQ AR degradation and suppressed toxicity of poyQ AR in Drosophila [59]. Similarly, Kazemi et al. by screening the fly genome for genes modulating the toxicity of polyglutamine, predicted dHDJ1, homologous to human Hsp40/HDJ1, and dTPR2, homologous to the human tetratricopeptide repeat protein 2 (TPR2) suppressed the toxicity of polyQ aggregates and verified in Drosophila models of polyQ diseases [49]. In addition, Y Kuo el at further demonstrated that co-expression of the HSP40 family protein DNAJ-1 and Hsp110 family protein, 70 kDa heat-shock cognate protein cb (HSC70cb), function together to suppress the cytotoxicity of mutated huntingtin in Drosophila HD model. Furthermore, DNAJB1, a human Hsp40, co-expressed with APG-1, a human Hsp110, in cells from HD Drosophila had a dramatic protective effect on polyQ-induced neural degeneration, whereas either component alone had little effect [60]. In our previous studies, using several types of SCA3/MJD Drosophila models, we have provided convincing proof that Hsp22 would be promising therapeutic agents with great potential against SCA3/MJD. Expression of MJDtr-Q78, a polyQ-expanded tract, showed significantly obvious SCA3/MJD phenotype including dramatic neurodegeneration, and completely faded pigmentation in adult flies with black point-like necrosis. Drosophila co-expressed polyQ-expanded protein together with either one or two copies of HSP22 gene intervened by heat shock, leading to differing corresponding mRNA levels mainly depending on the induced number of HSP22 gene copies. Findings suggested that Hsp22 showed positive influence on eye depigmentation (Figure 2), growth restriction, ability for eclosion and median lifespan [61].

SCA3/MJD Drosophila that expresses polyQ-expanded tracts in fly compound eyes. Overexpression of Hsp22, VPA and LiCl rescues polyQ-induced eye depigmentation in Drosophila model of SCA3/MJD. (A) Hsp22, (B) VPA, (C) LiCl. Paired images of adult fly eyes are showing, dissecting microscope (A, ×65; B, ×115; C, ×80) and electron microscope (×1000).
Acceleration on degradation of toxic proteins
Stimulation of cellular protein degradation mechanisms that target misfolded causative mutant proteins might be potential against polyQ diseases. Activation of autophagy and several small molecules presented positive influence on disease proteins clearance. By stimulating autophagy, rapamycin, the mTOR inhibitor, suppressed neurodegeneration in cell, Drosophila and mouse disease models [62, 85, 86] However, the various deleterious side-effects including dysfunction of kidney and lung, increased risk of infection and hyperlipidemia, prevent it from widely using in polyQ diseases [63–66]. In HD Drosophila model, lithium (Li) acting through the Wnt/Wg pathway, as a glycogen synthase kinase-3 (GSK3) β-specific inhibitor, showed protective effects against the toxicity caused by aggregates of causative polyQ proteins [87]. Similarly, since Li plays neuroprotective role in numerous models of neurodegenerative disorders and could induce autophagy for reducing the mutant protein aggregates, in our previous study, we confirmed its neuroprotective effects against protein toxicity induced by polyQ repeat expansion. As SCA3/MJD is the most common spinocerebellar ataxia in minland China [70]. Adenosine 3′, 5′-monophosphate (cAMP) response element–binding protein (CREB)–binding protein (CBP) [101, 102], a histone acetyltransferase (HAT) found in polyQ inclusions, its decreased activity contributes to polyQ disease. In SCA3/MJD Drosophila model, trichostatin A (TSA) protected against the increased rate of repeat instability, by compensating for decreased Drosophila CBP and/or HAT activity [71]. Additionally, we utilized HDAC inhibitor valproic acid (VPA) [72], a potential therapeutic agent, in Drosophila SCA3/MJD model. Deleterious phenotypes, including faded eye pigmentation, decreased climbing ability and shortened average life span, were similar to characteristics of SCA3/MJD patients. To examine the therapeutic effects of VPA in vivo, daily doses of VPA at various levels were administered to SCA3/MJD Drosophila before cross-breeding. We suggested that chronic treatment with VPA at optimum dose partly arrested eye depigmentation (Figure 2), ameliorated climbing disability, and prolonged the average lifespan of SCA3/MJD transgenic Drosophila.
Additionally, present researches direct to detect genetic modifiers that rescue polyQ degeneration, thus provide a novel target for testing therapeutic strategies. Recenly, Yamanaka et al applied RNAi screening in mouse neuro2a cells to identify modifiers for mutant huntingtin aggregates, and identified 111 shRNAs including 63 shRNAs suppressing huntingtin aggregation and 48 shRNAs performing an opposite effect [103]. We look forward to further study on testing neuroprotective effects of these 63 shRNAs for HD in vivo. Another study on genetic modifier in SCA3/MJD model of Drosophila confirmed that up-regulation of dMyc, a homologue of human cMyc proto-oncogene, could restore morphology and functional vision of eyes and suppress lethality by increased cellular level of CBP, which subsequently improve the status of histone acetylation and finally inhibit the compiling of polyQ aggregates [73].
Amelioration of mitochondrial dysfunctions
Compared with polyQ disease proteins, expanded CAG mutant transcripts are different polyQ pathogenic species to induce neurodegeneration [104]. Nucleolar stress is a p53-mediated pathway through which the nucleolus communicates with mitochondria to induce apoptosis by down-regulation of ribosomal RNA (rRNA) expression [105]. The nucleolar stress is activated by the expanded CAG mutant transcripts interacting directly with nucleolin (NCL), an essential protein for rRNA transcription, resulting in down-regulation of NCL binding to upstream control element (UCE) of the rRNA promoter [106], subsequently, UCE DNA CpG hypermethylation and the down-regulation of rRNA transcription [104]. Consequently, apoptosis is provoked by p53 protein accumulating in the mitochondria, which causes cytochrome c release and caspase activation [106]. Additionally, evidence that mitochondrial dysfunction is correlated with HD pathogenesis has been presented, by identified N-terminal expanded htt on neuronal mitochondrial membranes through electron microscopy [27]. According to these, numerous biological molecules and pharmacological compounds have shown efficacy in treatment for polyQ diseases, particularly for HD. Mitochondrial uncoupling proteins (UCPs), anion-carrier proteins located in the inner membrane of mitochondrial, modulate ATP and reactive oxygen species in mitochondria. Although UCPs failed to prevent the HD toxicity in neurons, by using genetic approaches in Drosophila, up-regulation of UCPs alleviated the HD locomotor behavior and early death of Drosophila when mHtt was selectively expressed in glia [74]. Additionally, inhibition of transglutaminase 2 (TG2), a transcriptional co-repressor, not only corrects transcriptional dysregulation in HD but also normalizes expression of mitochondrial genes, and protect neurons from excitotoxicity [107]. Meclizine, a clinically used drug that has correlated with mitochondrial respiration suppression, in Drosophila melanogaster models of HD, played a neuroprotective role against neuronal dystrophy and cell death, implicating that meclizine, capable of crossing the blood–brain barrier, may hold therapeutic potential against HD [75].
Conclusion
To date, targeting the pathogenic mechanisms, the fact that various therapeutic strategies have shown protective efficacy in Drosophila models of polyQ diseases gives new insight to researchers and clinical doctors as well as presents hope for patients of these diseases. Unfortunately, only a few of these therapeutic strategies have been testing in clinical trial, and the clinical efficacy still illusive. Anyhow, as Drosophila melanogaster presents various advantages and convenience, we can anticipate that in the future more therapeutic targets will be discovered by further understanding the pathogenic mechanisms on polyQ diseases, thus increasingly biological molecules and pharmacological compounds for polyQ diseases might be tested by employing Drosophila models, and these models of polyQ diseases could provide diverse options and clues for clinical application.
Ethical approval
The study was approved by the Expert Committee of **angya Hospital of Central South University in China (equivalent to an Institutional Review Board).
Abbreviations
- polyQ:
-
Polyglutamine
- SBMA:
-
Spinal bulbar muscular atrophy
- AR:
-
Androgen receptor
- SCA:
-
Spinocerebellar ataxias
- DRPLA:
-
Dentatorubral-pallidoluysian atrophy
- HD:
-
Huntington disease
- HDL2:
-
Huntington disease-like 2
- ADCAs:
-
Autosomal dominant cerebellar ataxias
- RNAi:
-
RNA interference
- MB:
-
Methylene blue
- tTG:
-
Tissue transglutaminase
- Htt:
-
Huntingtin
- intrabody:
-
Intracellular antibody
- QBP1:
-
Polyglutamine binding peptide 1
- Hsp:
-
Heat shock protein
- TPR2:
-
Tetratricopeptide repeat protein 2
- HSC70cb:
-
70 kDa heat-shock cognate protein cb
- Li:
-
Lithium
- GSK3:
-
Glycogen synthase kinase-3
- LiCl:
-
Lithium chloride
- HDAC:
-
Histone deacetylase
- SAHA:
-
Suberoylanilide hydroxamic acid
- TSA:
-
Trichostatin A
- CBP:
-
Adenosine 3′, 5′-monophosphate (cAMP) response element–binding protein (CREB)–binding protein
- HAT:
-
Histone acetyltransferase
- VPA:
-
Valproic acid
- rRNA:
-
Ribosomal RNA
- NCL:
-
Nucleolin
- UCE:
-
Upstream control element
- UCPs:
-
Uncoupling proteins
- TG2:
-
Transglutaminase 2.
References
Costa MC, Paulson HL: Toward understanding Machado-Joseph disease. Prog Neurobiol 2012, 97: 239–257.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH: Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352: 77–79.
Gatchel JR, Zoghbi HY: Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet 2005, 6: 743–755.
Shao J, Diamond MI: Polyglutamine diseases: emerging concepts in pathogenesis and therapy. Hum Mol Genet 2007, 16 Spec No. 2: R115-R123.
Zoghbi HY, Orr HT: Glutamine repeats and neurodegeneration. Annu Rev Neurosci 2000, 23: 217–247.
Paradisi I, Ikonomu V, Arias S: Huntington disease-like 2 (HDL2) in Venezuela: frequency and ethnic origin. J Hum Genet 2013, 58: 3–6.
Durr A: Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010, 9: 885–894.
Finsterer J: Perspectives of Kennedy’s disease. J Neurol Sci 2010, 298: 1–10.
Walker FO: Huntington’s disease. Lancet 2007, 369: 218–228.
Rubinsztein DC: Lessons from animal models of Huntington’s disease. Trends Genet 2002, 18: 202–209.
Ross CA: Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002, 35: 819–822.
Michalik A, Van Broeckhoven C: Pathogenesis of polyglutamine disorders: aggregation revisited. Hum Mol Genet 2003, 12 Spec No 2: R173-R186.
Difiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N: Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277: 1990–1993.
Ross CA, Tabrizi SJ: Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 2011, 10: 83–98.
Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL: Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet 1999, 8: 673–682.
Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE: Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell 2001, 12: 1393–1407.
Cummings CJ, Reinstein E, Sun Y, Antalffy B, Jiang Y, Ciechanover A, Orr HT, Beaudet AL, Zoghbi HY: Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron 1999, 24: 879–892.
Cummings CJ, Mancini MA, Antalffy B, Defranco DB, Orr HT, Zoghbi HY: Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 1998, 19: 148–154.
Okazawa H: Polyglutamine diseases: a transcription disorder? Cell Mol Life Sci 2003, 60: 1427–1439.
Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, Takiyama Y, Nishizawa M, Goto J, Kanazawa I, Davidson I, Tanese N, Takahashi H, Tsuji S: Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet 2000, 26: 29–36.
Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D: Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296: 2238–2243.
Nucifora FJ, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA: Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291: 2423–2428.
Mccampbell A, Taylor JP, Taye AA, Robitschek J, Li M, Walcott J, Merry D, Chai Y, Paulson H, Sobue G, Fischbeck KH: CREB-binding protein sequestration by expanded polyglutamine. Hum Mol Genet 2000, 9: 2197–2202.
Stenoien DL, Mielke M, Mancini MA: Intranuclear ataxin1 inclusions contain both fast- and slow-exchanging components. Nat Cell Biol 2002, 4: 806–810.
Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS: Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40: 25–40.
Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, Macdonald ME, Mcphaul MJ, Brady ST: Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 2003, 40: 41–52.
Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT: Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci 2002, 5: 731–736.
Browne SE, Bowling AC, Macgarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF: Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol 1997, 41: 646–653.
Ruan Q, Lesort M, Macdonald ME, Johnson GV: Striatal cells from mutant huntingtin knock-in mice are selectively vulnerable to mitochondrial complex II inhibitor-induced cell death through a non-apoptotic pathway. Hum Mol Genet 2004, 13: 669–681.
Orrenius S, Zhivotovsky B, Nicotera P: Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 2003, 4: 552–565.
Tang TS, Tu H, Chan EY, Maximov A, Wang Z, Wellington CL, Hayden MR, Bezprozvanny I: Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1. Neuron 2003, 39: 227–239.
Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinas R, Kristal BS, Hayden MR, Bezprozvanny I: Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc Natl Acad Sci U S A 2005, 102: 2602–2607.
Matsuyama Z, Yanagisawa NK, Aoki Y, Black JR, Lennon VA, Mori Y, Imoto K, Inuzuka T: Polyglutamine repeats of spinocerebellar ataxia 6 impair the cell-death-preventing effect of CaV2.1 Ca2+ channel–loss-of-function cellular model of SCA6. Neurobiol Dis 2004, 17: 198–204.
Bezprozvanny I, Hayden MR: Deranged neuronal calcium signaling and Huntington disease. Biochem Biophys Res Commun 2004, 322: 1310–1317.
Durr A, Stevanin G, Cancel G, Duyckaerts C, Abbas N, Didierjean O, Chneiweiss H, Benomar A, Lyon-Caen O, Julien J, Serdaru M, Penet C, Agid Y, Brice A: Spinocerebellar ataxia 3 and Machado-Joseph disease: clinical, molecular, and neuropathological features. Ann Neurol 1996, 39: 490–499.
Maciel P, Gaspar C, Destefano AL, Silveira I, Coutinho P, Radvany J, Dawson DM, Sudarsky L, Guimaraes J, Loureiro JE, Et A: Correlation between CAG repeat length and clinical features in Machado-Joseph disease. Am J Hum Genet 1995, 57: 54–61.
Ranum LP, Lundgren JK, Schut LJ, Ahrens MJ, Perlman S, Aita J, Bird TD, Gomez C, Orr HT: Spinocerebellar ataxia type 1 and Machado-Joseph disease: incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, or sporadic ataxia. Am J Hum Genet 1995, 57: 603–608.
Riess O, Rub U, Pastore A, Bauer P, Schols L: SCA3: neurological features, pathogenesis and animal models. Cerebellum 2008, 7: 125–137.
Venken KJ, Bellen HJ: Emerging technologies for gene manipulation in Drosophila melanogaster. Nat Rev Genet 2005, 6: 167–178.
Matthews KA, Kaufman TC, Gelbart WM: Research resources for Drosophila: the expanding universe. Nat Rev Genet 2005, 6: 179–193.
Phelps CB, Brand AH: Ectopic gene expression in Drosophila using GAL4 system. Methods 1998, 14: 367–379.
Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, Mccall A, Canal I, Orr HT, Zoghbi HY, Botas J: Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 2000, 408: 101–106.
Rubin GM, Yandell MD, Wortman JR, Gabor MG, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, Cherry JM, Henikoff S, Skupski MP, Misra S, Ashburner M, Birney E, Boguski MS, Brody T, Brokstein P, Celniker SE, Chervitz SA, Coates D, Cravchik A, Gabrielian A, Galle RF, Gelbart WM, George RA, Goldstein LS, Gong F, Guan P, et al.: Comparative genomics of the eukaryotes. Science 2000, 287: 2204–2215.
Bilen J, Bonini NM: Drosophila as a model for human neurodegenerative disease. Annu Rev Genet 2005, 39: 153–171.
Iijima-Ando K, Wu P, Drier EA, Iijima K, Yin JC: cAMP-response element-binding protein and heat-shock protein 70 additively suppress polyglutamine-mediated toxicity in Drosophila. Proc Natl Acad Sci U S A 2005, 102: 10261–10266.
Marsh JL, Walker H, Theisen H, Zhu YZ, Fielder T, Purcell J, Thompson LM: Expanded polyglutamine peptides alone are intrinsically cytotoxic and cause neurodegeneration in Drosophila. Hum Mol Genet 2000, 9: 13–25.
Taylor JP, Taye AA, Campbell C, Kazemi-Esfarjani P, Fischbeck KH, Min KT: Aberrant histone acetylation, altered transcription, and retinal degeneration in a Drosophila model of polyglutamine disease are rescued by CREB-binding protein. Genes Dev 2003, 17: 1463–1468.
Jackson GR, Salecker I, Dong X, Yao X, Arnheim N, Faber PW, Macdonald ME, Zipursky SL: Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998, 21: 633–642.
Kazemi-Esfarjani P, Benzer S: Genetic suppression of polyglutamine toxicity in Drosophila. Science 2000, 287: 1837–1840.
Mallik M, Lakhotia SC: RNAi for the large non-coding hsromega transcripts suppresses polyglutamine pathogenesis in Drosophila models. RNA Biol 2009, 6: 464–478.
Apostol BL, Kazantsev A, Raffioni S, Illes K, Pallos J, Bodai L, Slepko N, Bear JE, Gertler FB, Hersch S, Housman DE, Marsh JL, Thompson LM: A cell-based assay for aggregation inhibitors as therapeutics of polyglutamine-repeat disease and validation in Drosophila. Proc Natl Acad Sci U S A 2003, 100: 5950–5955.
Agrawal N, Pallos J, Slepko N, Apostol BL, Bodai L, Chang LW, Chiang AS, Thompson LM, Marsh JL: Identification of combinatorial drug regimens for treatment of Huntington’s disease using Drosophila. Proc Natl Acad Sci U S A 2005, 102: 3777–3781.
Bortvedt SF, Mclear JA, Messer A, Ahern-Rindell AJ, Wolfgang WJ: Cystamine and intrabody co-treatment confers additional benefits in a fly model of Huntington’s disease. Neurobiol Dis 2010, 40: 130–134.
Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM: Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington’s disease models. J Neurosci 2012, 32: 11109–11119.
Schulte J, Sepp KJ, Wu C, Hong P, Littleton JT: High-content chemical and RNAi screens for suppressors of neurotoxicity in a Huntington’s disease model. PLoS ONE 2011, 6: e23841.
Nagai Y, Fujikake N, Ohno K, Higashiyama H, Popiel HA, Rahadian J, Yamaguchi M, Strittmatter WJ, Burke JR, Toda T: Prevention of polyglutamine oligomerization and neurodegeneration by the peptide inhibitor QBP1 in Drosophila. Hum Mol Genet 2003, 12: 1253–1259.
Arribat Y, Bonneaud N, Talmat-Amar Y, Layalle S, Parmentier ML, Maschat F: A huntingtin peptide inhibits polyQ-huntingtin associated defects. PLoS ONE 2013, 8: e68775.
Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM: Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet 1999, 23: 425–428.
Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, Osawa Y, Collins CA, Gestwicki JE, Lieberman AP: Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol 2013, 9: 112–118.
Kuo Y, Ren S, Lao U, Edgar BA, Wang T: Suppression of polyglutamine protein toxicity by co-expression of a heat-shock protein 40 and a heat-shock protein 110. Cell Death Dis 2013, 4: e833.
Li Q, Jiang H, Yi J, Liao S, Shen L, Pan Q, **a K, Tang B: Research on Neuroprotective Role of Hsp22 in SCA3/MJD Transgenic Drosophila Models. Progr Biochem Biophys 2008, 35: 1430–1436.
Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O’Kane CJ, Rubinsztein DC: Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004, 36: 585–595.
Morrisett JD, Abdel-Fattah G, Hoogeveen R, Mitchell E, Ballantyne CM, Pownall HJ, Opekun AR, Jaffe JS, Oppermann S, Kahan BD: Effects of sirolimus on plasma lipids, lipoprotein levels, and fatty acid metabolism in renal transplant patients. J Lipid Res 2002, 43: 1170–1180.
Letavernier E, Bruneval P, Mandet C, Duong VHJ, Peraldi MN, Helal I, Noel LH, Legendre C: High sirolimus levels may induce focal segmental glomerulosclerosis de novo. Clin J Am Soc Nephrol 2007, 2: 326–333.
Kuypers DR: Benefit-risk assessment of sirolimus in renal transplantation. Drug Saf 2005, 28: 153–181.
Maroto JP, Hudes G, Dutcher JP, Logan TF, White CS, Krygowski M, Cincotta M, Shapiro M, Duran I, Berkenblit A: Drug-related pneumonitis in patients with advanced renal cell carcinoma treated with temsirolimus. J Clin Oncol 2011, 29: 1750–1756.
Jia DD, Zhang L, Chen Z, Wang CR, Huang FZ, Duan RH, **a K, Tang BS, Jiang H: Lithium chloride alleviates neurodegeneration partly by inhibiting activity of GSK3beta in a SCA3 Drosophila model. Cerebellum 2013, 12: 892–901.
Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, Bonini NM: Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell 2005, 18: 37–48.
Cushman-Nick M, Bonini NM, Shorter J: Hsp104 suppresses polyglutamine-induced degeneration post onset in a drosophila MJD/SCA3 model. PLoS Genet 2013, 9: e1003781.
Steffan JS, Bodai L, Pallos J, Poelman M, Mccampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM: Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature 2001, 413: 739–743.
Jung J, Bonini N: CREB-binding protein modulates repeat instability in a Drosophila model for polyQ disease. Science 2007, 315: 1857–1859.
Yi J, Zhang L, Tang B, Han W, Zhou Y, Chen Z, Jia D, Jiang H: Sodium valproate alleviates neurodegeneration in SCA3/MJD via suppressing apoptosis and rescuing the hypoacetylation levels of histone H3 and H4. PLoS ONE 2013, 8: e54792.
Singh MD, Raj K, Sarkar S: Drosophila Myc, a novel modifier suppresses the poly(Q) toxicity by modulating the level of CREB binding protein and histone acetylation. Neurobiol Dis 2014, 63: 48–61.
Besson MT, Dupont P, Fridell YW, Lievens JC: Increased energy metabolism rescues glia-induced pathology in a Drosophila model of Huntington’s disease. Hum Mol Genet 2010, 19: 3372–3382.
Gohil VM, Offner N, Walker JA, Sheth SA, Fossale E, Gusella JF, Macdonald ME, Neri C, Mootha VK: Meclizine is neuroprotective in models of Huntington’s disease. Hum Mol Genet 2011, 20: 294–300.
Koeppen AH: The pathogenesis of spinocerebellar ataxia. Cerebellum 2005, 4: 62–73.
Ross CA: Intranuclear neuronal inclusions: a common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 1997, 19: 1147–1150.
**a H, Mao Q, Eliason SL, Harper SQ, Martins IH, Orr HT, Paulson HL, Yang L, Kotin RM, Davidson BL: RNAi suppresses polyglutamine-induced neurodegeneration in a model of spinocerebellar ataxia. Nat Med 2004, 10: 816–820.
Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL: RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A 2005, 102: 5820–5825.
Wang AM, Morishima Y, Clapp KM, Peng HM, Pratt WB, Gestwicki JE, Osawa Y, Lieberman AP: Inhibition of hsp70 by methylene blue affects signaling protein function and ubiquitination and modulates polyglutamine protein degradation. J Biol Chem 2010, 285: 15714–15723.
van Bebber F, Paquet D, Hruscha A, Schmid B, Haass C: Methylene blue fails to inhibit Tau and polyglutamine protein dependent toxicity in zebrafish. Neurobiol Dis 2010, 39: 265–271.
Ciechanover A, Brundin P: The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron 2003, 40: 427–446.
Chan HY, Warrick JM, Gray-Board GL, Paulson HL, Bonini NM: Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet 2000, 9: 2811–2820.
Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY: Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet 2001, 10: 1511–1518.
Ravikumar B, Berger Z, Vacher C, O’Kane CJ, Rubinsztein DC: Rapamycin pre-treatment protects against apoptosis. Hum Mol Genet 2006, 15: 1209–1216.
Ravikumar B, Duden R, Rubinsztein DC: Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 2002, 11: 1107–1117.
Berger Z, Ttofi EK, Michel CH, Pasco MY, Tenant S, Rubinsztein DC, O’Kane CJ: Lithium rescues toxicity of aggregate-prone proteins in Drosophila by perturbing Wnt pathway. Hum Mol Genet 2005, 14: 3003–3011.
Tang B, Liu C, Shen L, Dai H, Pan Q, **g L, Ouyang S, **a J: Frequency of SCA1, SCA2, SCA3/MJD, SCA6, SCA7, and DRPLA CAG trinucleotide repeat expansion in patients with hereditary spinocerebellar ataxia from Chinese kindreds. Arch Neurol 2000, 57: 540–544.
Wang J, Shen L, Lei L, Xu Q, Zhou J, Liu Y, Guan W, Pan Q, **a K, Tang B, Jiang H: Spinocerebellar ataxias in mainland China: an updated genetic analysis among a large cohort of familial and sporadic cases. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2011, 36: 482–489.
Wang G, Sawai N, Kotliarova S, Kanazawa I, Nukina N: Ataxin-3, the MJD1 gene product, interacts with the two human homologs of yeast DNA repair protein RAD23, HHR23A and HHR23B. Hum Mol Genet 2000, 9: 1795–1803.
Wang Q, Li L, Ye Y: Regulation of retrotranslocation by p97-associated deubiquitinating enzyme ataxin-3. J Cell Biol 2006, 174: 963–971.
Doss-Pepe EW, Stenroos ES, Johnson WG, Madura K: Ataxin-3 interactions with rad23 and valosin-containing protein and its associations with ubiquitin chains and the proteasome are consistent with a role in ubiquitin-mediated proteolysis. Mol Cell Biol 2003, 23: 6469–6483.
Zhong X, Pittman RN: Ataxin-3 binds VCP/p97 and regulates retrotranslocation of ERAD substrates. Hum Mol Genet 2006, 15: 2409–2420.
Sanchez Y, Lindquist SL: HSP104 required for induced thermotolerance. Science 1990, 248: 1112–1115.
Parsell DA, Kowal AS, Singer MA, Lindquist S: Protein disaggregation mediated by heat-shock protein Hsp104. Nature 1994, 372: 475–478.
Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G: Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 2004, 13: 1183–1192.
Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP: Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A 2003, 100: 2041–2046.
Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM: Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci 2003, 23: 9418–9427.
Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF: Neuroprotective effects of phenylbutyrate in the N171–82Q transgenic mouse model of Huntington’s disease. J Biol Chem 2005, 280: 556–563.
Ying M, Xu R, Wu X, Zhu H, Zhuang Y, Han M, Xu T: Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem 2006, 281: 12580–12586.
Hasan S, Hottiger MO: Histone acetyl transferases: a role in DNA repair and DNA replication. J Mol Med (Berl) 2002, 80: 463–474.
Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P: Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Mol Cell 2002, 9: 265–277.
Yamanaka T, Wong HK, Tosaki A, Bauer PO, Wada K, Kurosawa M, Shimogori T, Hattori N, Nukina N: Large-scale RNA interference screening in Mammalian cells identifies novel regulators of mutant huntingtin aggregation. PLoS ONE 2014, 9: e93891.
Tsoi H, Chan HY: Roles of the nucleolus in the CAG RNA-mediated toxicity. Biochim Biophys Acta 1842, 2014: 779–784.
Zhang Y, Lu H: Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009, 16: 369–377.
Tsoi H, Lau TC, Tsang SY, Lau KF, Chan HY: CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci U S A 2012, 109: 13428–13433.
Mcconoughey SJ, Basso M, Niatsetskaya ZV, Sleiman SF, Smirnova NA, Langley BC, Mahishi L, Cooper AJ, Antonyak MA, Cerione RA, Li B, Starkov A, Chaturvedi RK, Beal MF, Coppola G, Geschwind DH, Ryu H, **a L, Iismaa SE, Pallos J, Pasternack R, Hils M, Fan J, Raymond LA, Marsh JL, Thompson LM, Ratan RR: Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med 2010, 2: 349–370.
Acknowledgements
The study was supported by the National Basic Research Program (973 Program) (No. 2012CB944601, 2012CB517902, 2011CB510002, to Hong Jiang), the New Century Excellent Talents in University (No. NCET-10-0836, to Hong Jiang), the National Natural Science Foundation of China (No. 81271260, 30971585 to Hong Jiang), Hunan Funds for Distinguished Young Scientists (No. 14JJ1008 to Hong Jiang), **njiang Natural Science Foundation (No.201318101-4 to Hong Jiang) and the Undergraduate Innovation Project of Central South University (No. CY12400 and YB13028 to Hong Jiang).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors have reported no actual or potential competing interest.
Authors’ contributions
HJ have made substantial contributions to conception and design; ZL have been involved in drafting the manuscript; BT have revised it critically for important intellectual content; HJ agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Long, Z., Tang, B. & Jiang, H. Alleviating neurodegeneration in Drosophila models of PolyQ diseases. cerebellum ataxias 1, 9 (2014). https://doi.org/10.1186/2053-8871-1-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/2053-8871-1-9