
Abstract
Background
Recently, array-comparative genomic hybridization (aCGH) platforms have significantly improved the resolution of chromosomal analysis allowing the identification of genomic copy number gains and losses smaller than 5 Mb. Here we report on a young man with unexplained severe mental retardation, autism spectrum disorder, congenital malformations comprising hypospadia and omphalocele, and episodes of high blood pressure. An ~ 6 Mb interstitial deletion that includes the causative genes is identified by oligonucleotide-based aCGH.
Results
Our index case exhibited a de novo chromosomal abnormality at 2q22 [del(2)(q22.1q22.3)dn] which was not visible at the 550 haploid band level. The deleted region includes eight genes: HNMT, SPOPL, NXPH2, LOC64702, LRP1B, KYNU, ARHGAP15 and GTDC1.
Discussion
aCGH revealed an ~ 6 Mb deletion in 2q22.1 to 2q22.3 in an as-yet unique clinical case associated with intellectual disability, congenital malformations and autism spectrum disorder. Interestingly, the deletion is co-localized with a fragile site (FRA2K), which could be involved in the formation of this chromosomal aberration. Further studies are needed to determine if deletions of 2q22.1 to 2q22.3 define a new microdeletion syndrome.
Similar content being viewed by others

Introduction
Intellectual disability (ID) or Mental Retardation (MR) affects a large number of individuals, and was recently estimated to have a prevalence of 1% in the general population [1]. Chromosomal abnormalities are causative factors in 4% to 34.1% of cases, detected mainly by G-band-based banding studies [2, 3]. The advent of array-comparative genomic hybridization (aCGH) has increased the detection rate by an additional 15–20% [4], generally by identifying submicroscopic chromosomal abnormalities. This progress has enabled a refined association of chromosomal aberrations and potentially underlying disease-causing genes, leading to better karyotype/genotype-phenotype correlations, and more qualified genetic counseling for families [5–8].
Genes associated with ID/MR can be found distributed throughout the human genome. According to the OMIM database [9] six genetic syndromes have been assigned to chromosomal region 2q22 to 2q23, including, Mowat-Wilson Syndrome (MWS) (MIM:235730); Nemaline Myopathy 2 (MIM:256030); Meier-Gorlin Syndrome 2 (MIM:613800); Susceptibility to Asthma (MIM:600807); Idiopathic Generalized Epilepsy 9 (MIM:607682); and, Hypogonadism, Alopecia, Diabetes Mellitus, Mental Retardation and Extrapyramidal Signs syndrome (MIM:241080). MWS is the best known disease in 2q22 ~ q23, presenting multiple congenital anomalies including Hirschsprung disease (HD) (MIM:142623) and MR. It has been recently associated with truncating mutations and/or heterozygous deletions of the ZEB2 homeobox gene (ZFHX1B) [10–13].
Here we present a Brazilian patient carrying a hitherto unreported ~ 6 Mb microdeletion in 2q22.1 to 2q22.3 upstream and outside the ZEB2 region [14]. The patient’s phenotype comprises severe autism spectrum disorder, associated to ID/MR, and congenital malformations, such as, omphalocele and hypospadia with cryptorchidism. Episodes of essential hypertension were an important feature in adolescence and were controlled with specific anti-hypertensive agents.
Case presentation
The patient is the only son of a young non-consanguineous couple, without any familial history. He was born at term by cesarean section; weight 3.950 g (>P50th percentile); length 51 cm (50th percentile). The patient had his first genetic evaluation at 4 years of age, being referred due to global developmental delay, lack of speech, an omphalocele (corrected by surgery), and balanic hypospadia with bilateral cryptorchidism. At physical examination he presented a coarse face with deep-set eyes, thick eyebrows, protruding tongue, small teeth, pointed chin, bulbous nose, wide spaced and hypoplastic nipples, scoliosis, corrected balanic hypospadia with a flattened gland, bilateral clinodactyly of the fifth finger, non-specific dermatoglyphic pattern, global developmental delay and behavioral disorder. A neurological evaluation through the Childhood Autism Rating Scale (CARS) protocol revealed a score of 44.5, compatible with an autism spectrum disorder.
Clinical follow-up proceeded on different occasions, and a series of hypertension episodes were detected when the patient was 17 years old, ranging from 150x100 mm Hg to 140x80 mm Hg. His body mass index (BMI) was 40 kg/m2, without any echocardiogram or electrocardiogram disturbance. Complete blood count, cranial cerebral tomography, thyroid hormones and biochemical evaluation were all normal, except for high triglycerides (289 mg/dl [normal values: 50–200 mg/dl]). He was treated with an angiotensin-converting enzyme inhibitor (captopril 25 mg daily) and dietetic measures, losing more than 40 kg in 2 years.
Recently, a new clinical evaluation was performed. He is currently 23 years old and presents in good physical health, with a BMI of 27.5 kg/m2 and normalization of his blood pressure without the use of antihypertensive drugs. He still suffers from a severe behavioral disorder, occasional tantrums, stereotyped movements of his trunk and repetitive whistling. Such clinical signs have improved dramatically with the use of antipsychotic drugs. Differential diagnoses such as Smith-Magenis, Simpson-Golabi-Behmel and Beckwith-Wiedemann syndromes have been ruled.
Results
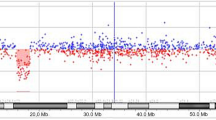
In the current case, G-banding and subtelomeric screening as well as molecular testing for the FMR1 and FMR2 genes were all normal (data not shown). aCGH identified an interstitial deletion of 6 Mb in the long arm of chromosome from 2q22.1 to 2q22.3, spanning positions 138,750,000 to 144,750,000 (Figure 1) and comprising eight genes (Table 1) [14]. The molecular cytogenetic karyotype according to ISCN 2009 was designated as: arr 2q22.1q22.3(138,750,000–144,750,000)x1. Seven out of 10 FISH probes used in the 2q22.1 band confirmed the deletion (Table 2). Eight to fourteen metaphase spreads were evaluated (Figure 2). The absence of xanthurenic acid in the patient’s urine showed that the metabolic pathway of tryptophan was not altered.

NimbleGen whole-genome oligonucleotide aCGH profile for chromosome 2q. The ideogram (grey bars) depicts 2q22.1 to 2q22.3 genomic regions with the cytogenetic bands on the long arm of chromosome 2 (not on scale). The ~ 6 Mb interstitial deletion interval at 2q22.1–q22.3 [hg19, chr2: 138,750,000–144,750,000 bp] is indicated by a red horizontal line below zero and the two black dotted vertical lines.

Three pairs of chromosome 2 are shown to illustrate the FISH results obtained with the BAC probes. a) RP11–587 K7 in 2q23.3 (green) and RP11–112 N16 in 2q22.1 (red) were located outside the deleted region. b) RP11–64O2 in 2q22.3 (green) is located outside the deleted region, while RP11–731 F1 in 2q22.1 (red) is within the affected region. c) RP11–137 J9 (green) and RP11–164E7 (red) both are missing on the derivative chromosome 2, indicating deletions in 2q22.1.
Human Genome Assembly Build 37 (hg19) shows that the region 2q22.1 to 2q22.3 is covered by small copy number variations, inversions (structural variations) and InDels, but no sequence gaps.
Discussion
A 23-year-old patient with ID/MR, autism, essential hypertension, and congenital malformations including an omphalocele and hypospadias with cryptorchidism is reported. He carries an ~6 Mb de novo microdeletion at 2q22.1–22.3 identified by an oligonucleotide aCGH panel [arr 2q22.1q22.3(138,750,000–144,750,000)x1 dn] according to Human Genome Assembly Build 37 (hg19).
Table 3 summarizes the clinical and genomic data from our index case and four patients described in the Decipher database with overlap** deletion intervals (138,750,000 to 144,750,000 bp) [17]. ID/MR is a common clinical feature, but none of the other listed cases presented with congenital malformations such as those found in our patient. Besides ID/MR, two patients show other common clinical features: strabismus and thick eyebrows (Patient 2566); and strabismus, bulbous nasal tip, and hypoplastic/inverted/absent nipples (Patient 1607). Interestingly, Patient 1607 has a complex karyotype involving chromosomes 2, 3 and 5 [18]. None of the individuals listed in Table 3 shared the exact breakpoints at 2q22.1–q22.3 as observed in our patient [17].
A child with MWS presenting with delayed psychomotor development, hypotonia, a variety of dysmorphic features, genitourinary anomalies and a severe course of HD has been described with a deletion at 2q22.2 to 2q22.3 [143,468,147–147,106,860] [19]. This 3.6 Mb aberration included ZEB2 and three other genes not currently associated with disease-KYNU, ARHGAP15 and GTDC1-all encoding for proteins involved in ubiquitous and non-specific pathways [20–22]. This deletion segment overlaps with our case in an ~ 1.2 Mb [143,468,147–144,750,000] comprising KYNU, ARHGAP15 and GTDC1 (Table 1). The authors speculate that those genes could play a crucial role in the process of tissue regeneration [19]. While many candidate genes have been studied to investigate their role in birth defects such as omphalocele and hypospadias/cryptorchidism [23–25], the clinical observations in our patient suggests the assignment of such malformations to the genes in the region 2q22.2–2q22.3.
Particularly interesting seems to be the function of the gene KYNU. It has previously been mentioned as possibly participating in a three-gene interaction influencing hypospadia, cryptorchidism and/or omphalocele [19]. However, a polymorphism in KYNU has also been linked to essential hypertension in a group of Han Chinese [34].
LRP1B is a very large human gene (1,9 Mb), located at 2q22.1 close to the fragile site, FRA2K, at 2q22.3 [36]. Many large genes residing within unstable chromosomal regions are highly evolutionarily conserved, and in general are not traditional mutational targets; however, genomic alterations can occur due to fragile site instability and contribute to diseases, including a variety of cancers [37]. Furthermore, there are important potential linkages between such genomic alterations and neurological development or neurodegeneration, for e.g. CNTNAP2 (2,3 Mb) localized within FRA7I at 7q35 found disrupted in a family with Gilles de la Tourette syndrome [38], and PARK2 (1.3 Mb) mutated in autosomal recessive juvenile Parkinson disease and located in the active center of FRA6E at 6q26 [39].
Fragile sites are understood to be specific loci that preferentially exhibit gaps and breaks in metaphase chromosomes following partial inhibition of DNA synthesis, and their break-prone areas are almost equally distributed along chromosomes [36, 40]. Human chromosome 2 has the highest number of fragile sites, with twenty one break-prone regions spaced at an average distance of 11.52 Mb [36]. The deletion studied in this report is placed at 2q22.1q22.3, overlap** FRA2K (2q22.3) and preceeded by FRA2F at 2q21.3. This is in line with the fact that regions of chromosomal instability at or near fragile sites are hot-spots for deletions and other alterations [41].
The fragile site neighboring 2q22.1q22.3 may have facilitated the chromosomal aberration in our patient including the entire LRP1B gene region. Moreover, a haploinsufficiency rank of 13.8% was recently established for this gene, just above the 0–10% range indicating a high likelihood of exhibiting haploinsufficiency [17]. If LRP1B is haploinsufficient in our patient, and considering its biological function within the central nervous system, it is tempting to speculate on the participation of this gene in the patient’s observed cognitive impairment. In addition, the presence of SNP variant rs2890652 (142,676,401) in LRP1B has been associated with BMI by genome wide association analysis [42]. While it is clear that correlation with potentially functional variants does not prove that these variants are causal, they can provide initial clues into which genes might be prioritized in further studies [42]. Consequently, LRP1B should be the subject of further studies to assign its relationship with BMI. The clinical management for hypertension and BMI in our patient at the age of 17 years included the use of an angiotensin-converting enzyme inhibitor and a vigilant diet.
Conclusion
Here we describe a patient presenting severe ID/MR, autism spectrum disorder, dysmorphism and congenital malformations, with episodes of high blood pressure associated with high levels of BMI. A whole-genome aCGH screening revealed an approximate 6 Mb de novo deletion, and a review of the literature provides indications of a new contiguous gene syndrome located in 2q22.1 to 2q22.3.
Methods
Peripheral blood chromosome analysis at the 550 G-band level was performed applying standard cytogenetic procedures. Molecular testing for FMR1 and FMR2 genes was performed [43, 44]; and the subtelomeric ToTel Vysion panel of probes (Abbott–Vysis) was also done. DNA from the patient was isolated from lymphocytes according to standard protocols and was subjected to aCGH analysis [Human Whole-Genome CGH; NimbleGen Systems, Madison, WI] to evaluate the presence of pathogenic copy number changes. The platform contained 385.000 oligonucleotides at a median spacing of 6 kb. The data was analyzed with the NimbleGen SignalMap v.1.9 software. Fluorescence in situ hybridization (FISH) using standard protocols with the following BAC clones as probes were used to confirm the deletion: RP11-112 N16, RP11-731 F1, RP11-597P14, RP11-231E19, RP11-137 J9, RP11-432O12, RP11-15D9, RP11-164E7, RP11-64O2, RP11-58 K7 (Table 2) [15, 16].
Urinary organic acids were analyzed to evaluate the metabolic pathway of tryptophan once the KYNU gene was found to be deleted inside the 2q22 region (Table 1). This biochemical analysis was performed by high-resolution gas chromatography coupled to mass spectrometry (Agilent 5975 C, HP-5).
The family consented to participate in the study, which was approved by the Brazilian Ethical Committee Board.
Consent
Written informed consent was obtained from the parents of the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- CARS:
-
Childhood Autism Rating Scale
- FISH:
-
Fluorescence in situ hybridization
- ID/MR:
-
Intellectual disability/Mental retardation
- ISCN:
-
International system for human cytogenetic nomenclature
- aCGH:
-
array-comparative genomic hybridization
- BMI:
-
Body Mass Index
- MWS:
-
Mowatt Wilson Syndrome
- HD:
-
Hirschprung disease.
References
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S: Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil 2008, 32(2):419–436.
Celep F, Sonmez FM, Karaguzel A: Chromosomal abnormalities in 457 Turkish patients with MCA/MR. Turk J Pediatr 2006, 48(2):130–134.
Xu J, Chen Z: Advances in molecular cytogenetics for the evaluation of mental retardation. Am J Med Genet C Semin Med Genet 2003, 117C(1):15–24. 10.1002/ajmg.c.10016
Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE, Epstein CJ, Faucett WA, Feuk L, Friedman JM, Hamosh A, Jackson L, Kaminsky EB, Kok K, Krantz ID, Kuhn RM, Lee C, Ostell JM, Rosenberg C, Scherer SW, Spinner NB, Stavropoulos DJ, Tepperberg JH, Thorland EC, Vermeesch JR, Waggoner DJ, Watson MS, Martin CL, Ledbetter DH: Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet 2010, 86: 749–764. 10.1016/j.ajhg.2010.04.006
Mulatinho M, Llerena J, Leren TP, Rao PN, Quintero-Rivera F: Deletion (1)(p32.2-p32.3) detected by array-CGH in a patient with developmental delay/mental retardation, dysmorphic features and low cholesterol: a new microdeletion syndrome? Am J Med Genet A 2008, 146A(17):2284–2290. 10.1002/ajmg.a.32454
Slavotinek AM: Novel microdeletion syndromes detected by chromosome microarrays. Hum Genet 2008, 124(1):1–17. 10.1007/s00439-008-0513-9
Ballif BC, Theisen A, McDonald-McGinn DM, Zackai EH, Hersh JH, Bejjani BA, Shaffer LG: Identification of a previously unrecognized microdeletion syndrome of 16q11.2q12.2. Clin Genet 2008, 74(5):469–475. 10.1111/j.1399-0004.2008.01094.x
Park SJ, Jung EH, Ryu RS, Kang HW, Ko JM, Kim HJ, Cheon CK, Hwang SH, Kang HY: Clinical implementation of whole-genome array CGH as a first-tier test in 5080 pre and postnatal cases. Mol Cytogenet 2011, 4: 12. 10.1186/1755-8166-4-12
OMIM. Online Mendelian Inheritance in Man http://www.ncbi.nlm.nih.gov/omim/, OMIM. Online Mendelian Inheritance in Man http://www.ncbi.nlm.nih.gov/omim/
Mowat DR, Croaker GD, Cass DT, Kerr BA, Chaitow J, Ades LC, Chia NL, Wilson MJ: Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J Med Genet 1998, 35(8):617–623. 10.1136/jmg.35.8.617
Amiel J, Lyonnet S: Hirschsprung disease, associated syndromes, and genetics: a review. J Med Genet 2001, 38(11):729–739. 10.1136/jmg.38.11.729
Zweier C, Albrecht B, Mitulla B, Behrens R, Beese M, Gillessen-Kaesbach G, Rott HD, Rauch A: “Mowat-Wilson” syndrome with and without Hirschsprung disease is a distinct, recognizable multiple congenital anomalies-mental retardation syndrome caused by mutations in the zinc finger homeo box 1B gene. Am J Med Genet 2002, 108(3):177–181. 10.1002/ajmg.10226
Zweier C, Temple IK, Beemer F, Zackai E, Lerman-Sagie T, Weschke B, Anderson CE, Rauch A: Characterisation of deletions of the ZFHX1B region and genotype-phenotype analysis in Mowat-Wilson syndrome. J Med Genet 2003, 40(8):601–605. 10.1136/jmg.40.8.601
Database of Genomic Variants. Genomic Variants in Human Genome GRCh 37:Feb.2009 (hg 19) http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg19/, Database of Genomic Variants. Genomic Variants in Human Genome GRCh 37:Feb.2009 (hg 19) http://projects.tcag.ca/cgi-bin/variation/gbrowse/hg19/
UCSC GENOME BIOINFORMATICS genome.ucsc.edu
CLONE DB http://www.ncbi.nlm.nih.gov/clone/, CLONE DB http://www.ncbi.nlm.nih.gov/clone/
DECIPHER database decipher.sanger.ac.uk
Hoffer MJ, Hilhorst-Hofstee Y, Knijnenburg J, Hansson KB, Engelberts AC, Laan LA, Bakker E, Rosenberg C: A 6 Mb deletion in band 2q22 due to a complex chromosome rearrangement associated with severe psychomotor retardation, microcephaly and distinctive dysmorphic facial features. Eur J Med Genet 2007, 50(2):149–154. 10.1016/j.ejmg.2006.11.004
Smigiel R, Szafranska A, Czyzewska M, Rauch A, Zweier C, Patkowski D: Severe clinical course of Hirschsprung disease in a Mowat-Wilson syndrome patient. J Appl Genet 2010, 51(1):111–113. 10.1007/BF03195718
Seoh ML, Ng CH, Yong J, Lim L, Leung T: ArhGAP15, a novel human RacGAP protein with GTPase binding property. FEBS Lett 2003, 539(1–3):131–137.
Zhao E, Li Y, Fu X, Zhang JY, Zeng H, Zeng L, Lin Y, Chen J, Yin G, Qian J, Ying K, **e Y, Zhao RC, Mao YM: Cloning and expression of human GTDC1 gene (glycosyltransferase-like domain containing 1) from human fetal library. DNA Cell Biol 2004, 23(3):183–187. 10.1089/104454904322964788
Christensen M, Duno M, Lund AM, Skovby F, Christensen E: Xanthurenic aciduria due to a mutation in KYNU encoding kynureninase. J Inherit Metab Dis 2007, 30(2):248–255. 10.1007/s10545-007-0396-2
Beleza-Meireles A, Lundberg F, Lagerstedt K, Zhou X, Omrani D, Frisen L, Nordenskjold A: FGFR2, FGF8, FGF10 and BMP7 as candidate genes for hypospadias. Eur J Hum Genet 2007, 15(4):405–410. 10.1038/sj.ejhg.5201777
Grati FR, Turolla L, D’Ajello P, Ruggeri A, Miozzo M, Bracalente G, Baldo D, Laurino L, Boldorini R, Frate E and others: Chromosome 11 segmental paternal isodisomy in amniocytes from two fetuses with omphalocoele: new highlights on phenotype-genotype correlations in Beckwith-Wiedemann syndrome. J Med Genet 2007, 44(4):257–263. 10.1136/jmg.2006.046854
Laitinen EM, Tommiska J, Virtanen HE, Oehlandt H, Koivu R, Vaaralahti K, Toppari J, Raivio T: Isolated cryptorchidism: no evidence for involvement of genes underlying isolated hypogonadotropic hypogonadism. Mol Cell Endocrinol 2011, 341(1–2):35–38.
Zhang Y, Zhang KX, He X, Yuan WT, Wang GL, Mao SY, Gao PJ, Huang W, Zhu DL: A polymorphism of kynureninase gene in a hypertensive candidate chromosomal region is associated with essential hypertension. Zhonghua **n Xue Guan Bing Za Zhi 2005, 33(7):588–591.
Mizutani K, Sugimoto K, Okuda T, Katsuya T, Miyata T, Tanabe T, Higaki J, Ogihara T, Yamori Y, Tsujita Y, Tago N, Iwai N: Kynureninase is a novel candidate gene for hypertension in spontaneously hypertensive rats. Hypertens Res 2002, 25(1):135–140. 10.1291/hypres.25.135
Kwok JB, Kapoor R, Gotoda T, Iwamoto Y, Iizuka Y, Yamada N, Isaacs KE, Kushwaha VV, Church WB, Schofield PR, Kapoor V: A missense mutation in kynurenine aminotransferase-1 in spontaneously hypertensive rats. J Biol Chem 2002, 277(39):35779–35782. 10.1074/jbc.C200303200
Komrower GM, Wilson V, Clamp JR, Westall RG: Hydroxykynureninuria: a Case of Abnormal Tryptophan Metabolism Probably Due to a Deficiency of Kynureninase. Arch Dis Child 1964, 39: 250–256. 10.1136/adc.39.205.250
Wang Y, O’Connell JR, McArdle PF, Wade JB, Dorff SE, Shah SJ, Shi X, Pan L, Rampersaud E, Shen H, Kim JD, Subramanya AR, Steinle NI, Parsa A, Ober CC, Welling PA, Chakravarti A, Weder AB, Cooper RS, Mitchell BD, Shuldiner AR, Chang YP: From the Cover: Whole-genome association study identifies STK39 as a hypertension susceptibility gene. Proc Natl Acad Sci U S A 2009, 106(1):226–231. 10.1073/pnas.0808358106
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, Aulchenko Y, Lumley T, Kottgen A, Vasan RS, Rivadeneira F, Eiriksdottir G, Guo X, Arking DE, Mitchell GF, Mattace-Raso FU, Smith AV, Taylor K, Scharpf RB, Hwang SJ, Sijbrands EJ, Bis J, Harris TB, Ganesh SK, O’Donnell CJ, Hofman A, Rotter JI, Coresh J, Benjamin EJ, Uitterlinden AG, Heiss G, Fox CS, Witteman JC, Boerwinkle E, Wang J, Gudnason V, Larson MG, Chakravarti A, Psaty BM, van Duijn CM: Genome-wide association study of blood pressure and hypertension. Nat Genet 2009, 41(6):677–687. 10.1038/ng.384
Aberg K, Dai F, Viali S, Tuitele J, Sun G, Indugula SR, Deka R, Weeks DE, McGarvey ST: Suggestive linkage detected for blood pressure related traits on 2q and 22q in the population on the Samoan islands. BMC Med Genet 2009, 10: 107. 10.1186/1471-2350-10-107
Cam JA, Zerbinatti CV, Knisely JM, Hecimovic S, Li Y, Bu G: The low density lipoprotein receptor-related protein 1B retains beta-amyloid precursor protein at the cell surface and reduces amyloid-beta peptide production. J Biol Chem 2004, 279(28):29639–29646. 10.1074/jbc.M313893200
Liu CX, Li Y, Obermoeller-McCormick LM, Schwartz AL, Bu G: The putative tumor suppressor LRP1B, a novel member of the low density lipoprotein (LDL) receptor family, exhibits both overlap** and distinct properties with the LDL receptor-related protein. J Biol Chem 2001, 276(31):28889–28896. 10.1074/jbc.M102727200
Liu CX, Musco S, Lisitsina NM, Yaklichkin SY, Lisitsyn NA: Genomic organization of a new candidate tumor suppressor gene, LRP1B. Genomics 2000, 69(2):271–274. 10.1006/geno.2000.6331
Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A: Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int J Oncol 2010, 36(4):929–940.
Smith DI, McAvoy S, Zhu Y, Perez DS: Large common fragile site genes and cancer. Semin Cancer Biol 2007, 17(1):31–41. 10.1016/j.semcancer.2006.10.003
Verkerk AJ, Mathews CA, Joosse M, Eussen BH, Heutink P, Oostra BA: CNTNAP2 is disrupted in a family with Gilles de la Tourette syndrome and obsessive compulsive disorder. Genomics 2003, 82(1):1–9. 10.1016/S0888-7543(03)00097-1
Denison SR, Callahan G, Becker NA, Phillips LA, Smith DI: Characterization of FRA6E and its potential role in autosomal recessive juvenile parkinsonism and ovarian cancer. Genes Chromosomes Cancer 2003, 38(1):40–52. 10.1002/gcc.10236
Durkin SG, Glover TW: Chromosome fragile sites. Annu Rev Genet 2007, 41: 169–192. 10.1146/annurev.genet.41.042007.165900
Lukusa T, Fryns JP: Human chromosome fragility. Biochim Biophys Acta 2008, 1779(1):3–16.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Magi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segre AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpelainen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, et al.: Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 2010, 42(11):937–948. 10.1038/ng.686
Haddad LA, Mingroni-Netto RC, Vianna-Morgante AM, Pena SD: A PCR-based test suitable for screening for fragile X syndrome among mentally retarded males. Hum Genet 1996, 97(6):808–812. 10.1007/BF02346194
Santos CB, Costa Lima MA, Pimentel MM: A new PCR assay useful for screening of FRAXE/FMR2 mental impairment among males. Hum Mutat 2001, 18(2):157–162. 10.1002/humu.1165
Acknowledgements
We are thankful for the patient’s family collaboration in this study, to Dr. Marcia Pimentel from SERVGEN, UERJ, for her kind help to allow us to use their molecular biology laboratory to perform FMR1 and FMR2 analysis. This work was partially supported by Brazilian grants: MVM received scholarships from CAPES (BEX:3692-05-1) and CNPq/FIOCRUZ (170060/2010-1) and JCLJr was granted by CNPq (Edital 061/2008; # 490754/2008-2). U19AI067769 grant (NR) from UCLA. Supported in part by the BMBF/DLR BRA 09/020.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interests.
Authors’ contributions
MVM drafted the manuscript, performed the cytogenetic analysis and the FISH subtelomeric screening, the molecular analysis for FMR1 and FMR2, and analyzed and interpreted the aCGH data. CLCS and JCLJr carried out clinical examination and evaluation of the patient. FS performed the biochemical analysis. DH and SP isolated and provided the BAC probes. KM performed the BAC-FISH relevant confirmation. AW, TL, NR and JCLJ coordinated the study. All authors improved and approved the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Mulatinho, M.V., de Carvalho Serao, C.L., Scalco, F. et al. Severe intellectual disability, omphalocele, hypospadia and high blood pressure associated to a deletion at 2q22.1q22.3: case report. Mol Cytogenet 5, 30 (2012). https://doi.org/10.1186/1755-8166-5-30
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-5-30