
Abstract
Background
In the biopharmaceutical industry, biomarkers define molecular taxonomies of patients and diseases and serve as surrogate endpoints in early-phase drug trials. Molecular biomarkers can be much more sensitive than traditional lab tests. Discriminating disease biomarkers by traditional method such as DNA microarray has proved challenging. Alternative splicing isoform represents a new class of diagnostic biomarkers. Recent scientific evidence is demonstrating that the differentiation and quantification of individual alternative splicing isoforms could improve insights into disease diagnosis and management. Identifying and characterizing alternative splicing isoforms are essential to the study of molecular mechanisms and early detection of complex diseases such as breast cancer. However, there are limitations with traditional methods used for alternative splicing isoform determination such as transcriptome-level, low level of coverage and poor focus on alternative splicing.
Results
Therefore, we presented a peptidomics approach to searching novel alternative splicing isoforms in clinical proteomics. Our results showed that the approach has significant potential in enabling discovery of new types of high-quality alternative splicing isoform biomarkers.
Conclusions
We developed a peptidomics approach for the proteomics community to analyze, identify, and characterize alternative splicing isoforms from MS-based proteomics experiments with more coverage and exclusive focus on alternative splicing. The approach can help generate novel hypotheses on molecular risk factors and molecular mechanisms of cancer in early stage, leading to identification of potentially highly specific alternative splicing isoform biomarkers for early detection of cancer.
Similar content being viewed by others
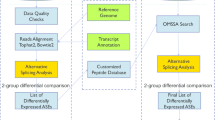

Introduction
A biomarker as defined by the National Cancer Institute is "a biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process, or of a condition or disease [1]." It is a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention [2]. The field of biomarkers has grown extensively over the past decade in many areas such as medicine, cell biology, genetics, geology and astrobiology, and ecotoxicology etc. and biomarkers are currently being studied in many academic centers and in industry.
In recent years, functional genomics studies using DNA Microarrays have been shown effective in identifying markers differentiating between breast cancer tissues and normal tissues, by measuring thousands of differentially expressed genes simultaneously [3–5].
However, early detection and treatment of breast cancer is still challenging. One reason is that obtaining tissue samples for microarray analysis can be still difficult. Another reason is that genes are not directly involved in any physical functions. On the contrary, the proteome, are the real functional molecules and the keys to understanding the development of cancer. Moreover, the fact that breast cancer is not a single homogeneous disease but consists of multiple disease status, each arising from a distinct molecular mechanism and having a distinct clinical progression path [6] makes the disease difficult to early detect.
Alternative splicing isoforms represent a new class of diagnostic biomarkers [7]. The chance of success with alternative splicing isoforms would be higher than the conventional approach [8, 9]. Alternative splicing occurs in 95% human genes and works by selecting specific exons and sometimes even intronic regions of the gene into mature mRNAs [10]. Alternative splicing accounts for approximately 8% of all protein isoforms which is any of several different forms of the same protein and have three types: alternative splicing, SNP, and posttranslational modification (PTM).
Recent scientific studies have shown that diseased cells may produce many types of splicing variants of common regulatory proteins, e.g., protein kinase C, 14-3-3, p53, and VGFR, which could provide novel insights into complex disease diagnosis and management, particularly in cancers [11–14]. Alternative mRNA splicing is an important source of achieving molecular functional diversity. It is often regulated in a temporal or tissue-specific fashion, giving rise to different protein isoforms in different tissues or developmental states mediated by extracellular signaling mechanisms [15, 16]. Splicing regulation is a key mechanism to tune gene expression to a variety of conditions and its dysfunction may often be at the basis of the onset of genetic disease and cancer [9]. In cancer, many examples of alternative splicing isoforms were reported [17–21]. For example, Julian et al. used a high-throughput reverse transcription-PCR-based system for splicing annotation to monitor the alternative splicing profiles of 600 cancer-associated genes in a panel of 21 normal and 26 cancerous breast tissues. They found that 41 alternative splicing events significantly differed in breast tumors relative to normal breast tissues and that most cancer-specific changes in splicing that disrupt known protein domains support an increase in cell proliferation or survival consistent with a functional role for alternative splicing in cancer. Compared to normal mRNA splicing events, alternative splicing mechanisms and patterns in complex diseases such as cancer can be quite complex. Finding alternative splicing isoforms or patterns of their development, therefore, have been promising in hel** develop high-quality biomarkers and targets for disease management [22].
Discovering disease biomarkers in the human plasma has been met with both enthusiasm and criticisms in recent years. On one hand, it is expected that disease conditions such as cancer may be diagnosed early by analyzing complex protein mixtures in easily accessible human blood (serum or plasma), in which proteins induced by cancer may be differentially cleaved, secreted, leaked out, and therefore differentially detected from normal healthy conditions. On the other hand, the bulk of proteins circulating in human blood vary in different conditions without cancers and across different individuals, and changes in protein expressions are often diluted in the blood -- extremely challenging for biological interpretation of protein quantification changes [23, 24]. The use of alternative splicing isoforms as potential biomarkers, therefore, offers a new opportunity to use the detectability instead of quantification of biomarker peptides, for the peptides that may map to unique alternative splicing isoforms specific to cancer.
However, systematic, comprehensive, proteome-scale experimental and computational characterization of protein isoforms directly at the protein/peptide level and with exclusive focus on alternative splicing has never been reported. Compared with the "indirect" transcriptome-level characterizations such as EST sequencing[Step 2: identification and characterization of alternative splicing isoform using proteomics The most popular three types of search algorithms are 1) correlating acquired MS/MS with theoretical spectrum, counts the number of peaks in common, such as: SEQUEST [37] and X!Tandem [38], 2) modeling the extent of peptide fragmentation, then estimates the probability that an assignment is incorrect due specifically to a random match, such as Mascot [39] and OMSSA [40], and 3) De Novo Sequencing such as Lutefisk[41] and PEAKS[42]. We first run OMSSA against SASD to identify peptide from MS data. Then we perform preliminary data analysis. Last, we extract information about alternative splicing. OMSSA reports hits ranked by E-value. An E-value for a hit is a score that is the expected number of random hits from a search library to a given spectrum, such that the random hits have an equal or better score than the hit. For example, a hit with an E-value of 1.0 implies that one hit with a score equal to or better than the hit being scored would be expected at random from a sequence library search [40]. The E-value is calculated to report the expected frequency of observing scores equivalent to or better than the one for the reported peptide if the results were to take place randomly. The lower the E-value is, the more significant the score for the identified peptide by the peptide search using SASD database is. One-sided Wilcoxon signed-rank test is used to perform the preliminary statistical analysis in order to identify peptides with significant occurrence differences in the health and breast cancer samples. We present two kinds of methods to validate isoforms in proteomics data, which are 1) literature curation of alternative splicing isoforms and 2) cross-validation of multiple studies. First, we perform an extensive literature curation to determine the constituents of alternative splicing isoform. Then, we validate results using independent proteomics datasets derived from other study. We believe that such an integrative systems approach is essential to development and validation of panel alternative splicing isoform that may withstand rigorous testing for the future steps. Pathway analysis is performed using the following databases: Integrated Pathway Analysis Database (IPAD) (http://bioinfo.hsc.unt.edu/ipad/) [43].Step 3: validation of alternative splicing isoform
Pathway analysis
Results
The 80 breast cancer plasma samples with 40 samples from women diagnosed with breast cancer and 40 from healthy volunteer women as controls were searched by OMSSA[40] against the SASD database. After OMSSA searching, preliminary statistical analysis, and alternative splicing, we identified the eight alternative splicing isoform biomarkers using the peptidomics approach to searching novel alternative splicing isoform in the proteomics data (Table 1). The peptides with E-value greater than 0.01 were filtered out. The P-value was calculated by performing one-sided Wilcoxon signed-rank test to examine the probability that the median difference between two groups of samples is greater than zero. The number of such peptide found in Health (h) and Cancer (c) samples are listed separately in the table. The tested peptides are more likely to exist in cancer samples than in healthy samples when P-value is small. Bold text is the left part of the junction and italic text is the right part. Splicing site is marked by ^ or (). '()' means the splicing site is shared by the left region and right region. For example, the second peptide QTPKHISESLGAEVDPDMSWSSSLATPPTLSSTVLI(G)LLHSSVK is a synthetic product of the ENST00000380152 in gene BRCA2 when its eighth, ninth, and tenth exons are skipped and its seventh exon is combined together with its eleventh exon. The Glycine is the shared splicing site between the seventh exon and the eleventh exon.
None of the eight peptides are reported to have ever been detected in the Peptide Atlas database, which contains a comprehensive catalogue of all peptides derived from published proteomics experiments. They are not found with normal splicing mechanism (Table 1).
The first sequence is a single intron alternative splicing. It was observed in 20 patient samples and 4 healthy samples. Triple play mode of the annotated Thermo-Finnegan LCQ-DECA ion-trap MS/MS spectrum is shown in Figure 2[44]. The triple play mode includes a) primary mass spectrum; b) zoom scan mass spectrum; c) MS/MS mass spectrum and d) protein identification from MS/MS). Due to space limit, the spectrums of other seven alternative splicing sequences are omitted.

Triple play mode spectrum of SWGGRPQRMGAVPGGVWSAVLMGGAR in patient C_024.
The eight peptides, identified by OMSSA, have significant difference in the numbers of hit samples between healthy women and breast cancers (pvalue < 0.05, Table 1). A screen shot from the UCSC genome browser [45] in the region of these peptides are also shown in Figure 3. It shows that these peptide sequences are not found in EST sequences and mRNA from Genbank and one refseq gene(Figure 3).

UCSC genome browser screen shot of genomic region for the novel peptide.
Table 1 shows that our peptidomics approach has significant potential in enabling discovery of new types of high-quality alternative splicing isoform biomarkers. Further literature search found that there are no any literature reports for the eight peptides. Moreover, a cross-validation found that identification of the eight peptides is supported by the independent Study which contains 40 samples collected from women diagnosed with breast cancer and 40 from healthy volunteer woman who served as controls.
Pathway analysis shows the pathways linked with the eight alternative splicing isoforms are transcription factor, signaling, cancer, and synthesis (Table 2).
Discussions
We described the peptidomics approach to searching novel alternative splicing isoform in proteomics data, especially artificial alternative splicing and SNP. We can use it to identify two types of common alternative splicing events: Exon Skip** and Intron Retention. Exon Skip** is an alternative splicing mechanism in which exon(s) are included or excluded from the final gene transcript leading to extended or shortened mRNA variants. And Intron Retention is an event in which an intron is retained in the final transcript. Other types of alternative splicing events such as alternative 3' splice site and 5' splice site are not included in our method but can be derived indirectly from the two basic types: exon skip** and intron retention.
The current protein sequence databases used by tandem mass spectra search engines, for example IPI, UniProt, and NCBI nr, are designed to be useful as possible to as many researchers as possible. As such, they are a less than ideal substrate for tandem mass spectra search. Protein sequence databases typically represent only "full-length" protein sequences and attempt to collapse protein variants to a single "consensus" entry. Tandem mass spectra search engines, however, chop up the protein sequence using an in-silico enzymatic digestion (such as trypsin), so full-length proteins are not needed in order to identify experimentally observed peptides; and the currently available search engines require the experimental peptides' sequences be explicitly present in the sequence database in order to identify them, so explicit sequence variants are very important. The SASD is in fact a complete peptide sequence database, which includes majority of all occurrence of alternative splicing. It also provides alternative splicing for each peptide, such as splicing mode, splicing type, splicing site, starting position, ending position, and peptide sequence.
Moreover, the current protein sequence databases and some alternative splicing database such as ASTD and EID are not ideal or enough for identifying alternative splicing isoform from tandem mass spectrometry. There are either no or very few isoform information in these databases. For example, ASTD only includes 9757 occurrences of intron isoforms and 5214 occurrences of exon isoforms. Even for Cassette exons event, the number of occurrences is only 12470[15]. In contrast, the SASD database in our method includes 11,919,779 Alternative Splicing peptides covering about 56,630 genes (ensembl gene IDs), 95,260 transcripts (ensembl transcript IDs), 1956 pathways, 6704 diseases, 5615 drugs, and 52 organs.
Its comprehensive coverage means better sensitivity in identifying novel alternative splicing isoforms than the PEPPI. And its exclusive focus on alternative splicing can definitely increase the specificity of the identification of alternative splicing.
Alternative splicing isoform biomarkers are apparently important and can serve as an alternative to traditional biomarkers. We can use quantitative information such as p-value to determine the significance of the marker. We can also use the qualitative information such as: splicing type, splicing mode, peptide sequence etc. to further analyze the alternative splicing's mechanism. We think that combination of traditional biomarkers with the alternative splicing isoform biomarkers will definitely help us better understand the treatment, diagnosis, and prognosis of cancer.
References
National Cancer Institute Dictionary.
Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clinical pharmacology and therapeutics. 2001, 69 (3): 89-95.
Hu X, Zhang Y, Zhang A, Li Y, Zhu Z, Shao Z, Zeng R, Xu LX: Comparative serum proteome analysis of human lymph node negative/positive invasive ductal carcinoma of the breast and benign breast disease controls via label-free semiquantitative shotgun technology. OMICS. 2009, 13 (4): 291-300. 10.1089/omi.2009.0016.
Zeidan BA, Cutress RI, Murray N, Coulton GR, Hastie C, Packham G, Townsend PA: Proteomic analysis of archival breast cancer serum. Cancer Genomics Proteomics. 2009, 6 (3): 141-147.
Lebrecht A, Boehm D, Schmidt M, Koelbl H, Schwirz RL, Grus FH: Diagnosis of breast cancer by tear proteomic pattern. Cancer Genomics Proteomics. 2009, 6 (3): 177-182.
Polyak K: Breast cancer: origins and evolution. J Clin Invest. 2007, 117 (11): 3155-3163. 10.1172/JCI33295.
Roberts TK, Eugenin EA, Morgello S, Clements JE, Zink MC, Berman JW: PrPC, the cellular isoform of the human prion protein, is a novel biomarker of HIV-associated neurocognitive impairment and mediates neuroinflammation. Am J Pathol. 2010, 177 (4): 1848-1860. 10.2353/ajpath.2010.091006.
Brinkman BM: Splice variants as cancer biomarkers. Clinical biochemistry. 2004, 37 (7): 584-594. 10.1016/j.clinbiochem.2004.05.015.
Garcia-Blanco MA, Baraniak AP, Lasda EL: Alternative splicing in disease and therapy. Nat Biotechnol. 2004, 22 (5): 535-546. 10.1038/nbt964.
Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ: Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nature reviews Molecular cell biology. 2013, 14 (3): 153-165.
Anensen N, Oyan AM, Bourdon JC, Kalland KH, Bruserud O, Gjertsen BT: A distinct p53 protein isoform signature reflects the onset of induction chemotherapy for acute myeloid leukemia. Clin Cancer Res. 2006, 12 (13): 3985-3992. 10.1158/1078-0432.CCR-05-1970.
Assender JW, Gee JM, Lewis I, Ellis IO, Robertson JF, Nicholson RI: Protein kinase C isoform expression as a predictor of disease outcome on endocrine therapy in breast cancer. J Clin Pathol. 2007, 60 (11): 1216-1221. 10.1136/jcp.2006.041616.
Cressey R, Wattananupong O, Lertprasertsuke N, Vinitketkumnuen U: Alteration of protein expression pattern of vascular endothelial growth factor(VEGF) from soluble to cell-associated isoform during tumourigenesis. BMC Cancer. 2005, 5: 128-10.1186/1471-2407-5-128.
Di Fede G, Giaccone G, Limido L, Mangieri M, Suardi S, Puoti G, Morbin M, Mazzoleni G, Ghetti B, Tagliavini F: The epsilon isoform of 14-3-3 protein is a component of the prion protein amyloid deposits of Gerstmann-Straussler-Scheinker disease. J Neuropathol Exp Neurol. 2007, 66 (2): 124-130. 10.1097/nen.0b013e3180302060.
Thanaraj TA, Stamm S, Clark F, Riethoven JJ, Le Texier V, Muilu J: ASD: the Alternative Splicing Database. Nucleic Acids Res. 2004, 32 (Database): D64-69.
Matter N, Herrlich P, Konig H: Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002, 420 (6916): 691-695. 10.1038/nature01153.
Lixia M, Zhijian C, Chao S, Chaojiang G, Congyi Z: Alternative splicing of breast cancer associated gene BRCA1 from breast cancer cell line. J Biochem Mol Biol. 2007, 40 (1): 15-21. 10.5483/BMBRep.2007.40.1.015.
Zhu Z, **ng S, Cheng P, Zeng F, Lu G: Modification of alternative splicing of Bcl-x pre-mRNA in bladder cancer cells. J Huazhong Univ Sci Technolog Med Sci. 2006, 26 (2): 213-216. 10.1007/BF02895819.
Ku TH, Hsu FR: Mining colon cancer specific alternative splicing in EST database. AMIA Annu Symp Proc. 2005, 1012-
Ogawa T, Shiga K, Hashimoto S, Kobayashi T, Horii A, Furukawa T: APAF-1-ALT, a novel alternative splicing form of APAF-1, potentially causes impeded ability of undergoing DNA damage-induced apoptosis in the LNCaP human prostate cancer cell line. Biochem Biophys Res Commun. 2003, 306 (2): 537-543. 10.1016/S0006-291X(03)00995-1.
Venables JP, Klinck R, Bramard A, Inkel L, Dufresne-Martin G, Koh C, Gervais-Bird J, Lapointe E, Froehlich U, Durand M, et al: Identification of alternative splicing markers for breast cancer. Cancer Res. 2008, 68 (22): 9525-9531. 10.1158/0008-5472.CAN-08-1769.
Skotheim RI, Nees M: Alternative splicing in cancer: noise, functional, or systematic?. Int J Biochem Cell Biol. 2007, 39 (7-8): 1432-1449. 10.1016/j.biocel.2007.02.016.
Rifai N, Gillette MA, Carr SA: Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006, 24 (8): 971-983. 10.1038/nbt1235.
Kulasingam V, Diamandis EP: Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nature clinical practice Oncology. 2008, 5 (10): 588-599. 10.1038/ncponc1187.
Adams MD, Kelley JM, Gocayne JD, Dubnick M, Polymeropoulos MH, **ao H, Merril CR, Wu A, Olde B, Moreno RF: Complementary DNA sequencing: expressed sequence tags and human genome project. Science. 1991, 252 (5013): 1651-1656. 10.1126/science.2047873.
Kapur K, **ng Y, Ouyang Z, Wong WH: Exon arrays provide accurate assessments of gene expression. Genome Biol. 2007, 8 (5): R82-10.1186/gb-2007-8-5-r82.
Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD: Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003, 302 (5653): 2141-2144. 10.1126/science.1090100.
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ: Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008, 40 (12): 1413-1415. 10.1038/ng.259.
Pando MP, Kotraiah V, McGowan K, Bracco L, Einstein R: Alternative isoform discrimination by the next generation of expression profiling microarrays. Expert Opin Ther Targets. 2006, 10 (4): 613-625. 10.1517/14728222.10.4.613.
Lee PS, Shaw LB, Choe LH, Mehra A, Hatzimanikatis V, Lee KH: Insights into the relation between mrna and protein expression patterns: II. Experimental observations in Escherichia coli. Biotechnol Bioeng. 2003, 84 (7): 834-841. 10.1002/bit.10841.
Mehra A, Lee KH, Hatzimanikatis V: Insights into the relation between mRNA and protein expression patterns: I. Theoretical considerations. Biotechnol Bioeng. 2003, 84 (7): 822-833. 10.1002/bit.10860.
Zhou A, Zhang F, Chen JY: PEPPI: a peptidomic database of human protein isoforms for proteomics experiments. BMC bioinformatics. 2010, S7-11 Suppl 6
Higgs RE, Knierman MD, Gelfanova V, Butler JP, Hale JE: Comprehensive label-free method for the relative quantification of proteins from biological samples. J Proteome Res. 2005, 4 (4): 1442-1450. 10.1021/pr050109b.
Zhang F, Drabier R: SASD: the Synthetic Alternative Splicing Database for identifying novel isoform from proteomics. BMC bioinformatics. 2013, S13-14 Suppl 14
Zhang F, Drabier R: IPAD: the Integrated Pathway Analysis Database for Systematic Enrichment Analysis. BMC bioinformatics. 2012, S7-13 Suppl 15
Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al: Ensembl 2013. Nucleic Acids Res. 2013, 41 (Database): D48-55.
Eng JK, McCormack AL, Yates Iii JR: An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. Journal of the American Society for Mass Spectrometry. 1994, 5 (11): 976-989. 10.1016/1044-0305(94)80016-2.
Bjornson RD, Carriero NJ, Colangelo C, Shifman M, Cheung KH, Miller PL, Williams K: X!!Tandem, an improved method for running X!tandem in parallel on collections of commodity computers. J Proteome Res. 2008, 7 (1): 293-299. 10.1021/pr0701198.
Koenig T, Menze BH, Kirchner M, Monigatti F, Parker KC, Patterson T, Steen JJ, Hamprecht FA, Steen H: Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J Proteome Res. 2008, 7 (9): 3708-3717. 10.1021/pr700859x.
Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH: Open mass spectrometry search algorithm. J Proteome Res. 2004, 3 (5): 958-964. 10.1021/pr0499491.
Johnson RS, Taylor JA: Searching sequence databases via de novo peptide sequencing by tandem mass spectrometry. Mol Biotechnol. 2002, 22 (3): 301-315. 10.1385/MB:22:3:301.
Ma B, Zhang K, Hendrie C, Liang C, Li M, Doherty-Kirby A, Lajoie G: PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid Commun Mass Spectrom. 2003, 17 (20): 2337-2342. 10.1002/rcm.1196.
Zhang F, Drabier R: IPAD: the Integrated Pathway Analysis Database for Systematic Enrichment Analysis. BMC Bioinformatics. 2012, 13 (14):
Wang M, Witzmann FA, Lyubimov AV: Proteomics in Drug Discovery and Development. Encyclopedia of Drug Metabolism and Interactions. 2011, John Wiley & Sons, Inc
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D: The human genome browser at UCSC. Genome Res. 2002, 12 (6): 996-1006.
Acknowledgements
We thank Hoosier Oncology Group for collecting breast cancer plasma samples. The proteomics study for biomarker discovery was supported by the National Cancer Institute Clinical Proteomics Technology Assessment for Cancer program (U24 CA126480). We also thank the support of Bioinformatics Program at University of North Texas Health Science Center.
Declarations
The publication costs for this article were funded by the bioinformatics program in University of North Texas Health Science Center.
This article has been published as part of BMC Systems Biology Volume 7 Supplement 5, 2013: Selected articles from the International Conference on Intelligent Biology and Medicine (ICIBM 2013): Systems Biology. The full contents of the supplement are available online at http://www.biomedcentral.com/bmcsystbiol/supplements/7/S5.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests statement
The authors declare that they have no competing interests.
Authors' contributions
RD and FZ conceived the initial work and designed the method. FZ and RD developed the alternative splicing database and method, and performed the computational analyses. MW provided experimental data. TM performed literature search. All authors are involved in the drafting and revisions of the manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhang, F., Wang, M., Michael, T. et al. Novel alternative splicing isoform biomarkers identification from high-throughput plasma proteomics profiling of breast cancer. BMC Syst Biol 7 (Suppl 5), S8 (2013). https://doi.org/10.1186/1752-0509-7-S5-S8
Published:
DOI: https://doi.org/10.1186/1752-0509-7-S5-S8