
Abstract
Background
Cytochrome P450 proteins (CYPs) play diverse and pivotal roles in fungal metabolism and adaptation to specific ecological niches. Fungal genomes encode extremely variable “CYPomes” ranging from one to more than 300 CYPs. Despite the rapid growth of sequenced fungal and oomycete genomes and the resulting influx of predicted CYPs, the vast majority of CYPs remain functionally uncharacterized. To facilitate the curation and functional and evolutionary studies of CYPs, we previously developed Fungal Cytochrome P450 Database (FCPD), which included CYPs from 70 fungal and oomycete species. Here we present a new version of FCPD (1.2) with more data and an improved classification scheme.
Results
The new database contains 22,940 CYPs from 213 species divided into 2,579 clusters and 115 clans. By optimizing the clustering pipeline, we were able to uncover 36 novel clans and to assign 153 orphan CYP families to specific clans. To augment their functional annotation, CYP clusters were mapped to David Nelson’s P450 databases, which archive a total of 12,500 manually curated CYPs. Additionally, over 150 clusters were functionally classified based on sequence similarity to experimentally characterized CYPs. Comparative analysis of fungal and oomycete CYPomes revealed cases of both extreme expansion and contraction. The most dramatic expansions in fungi were observed in clans CYP58 and CYP68 (Pezizomycotina), clans CYP5150 and CYP63 (Agaricomycotina), and family CYP509 (Mucoromycotina). Although much of the extraordinary diversity of the pan-fungal CYPome can be attributed to gene duplication and adaptive divergence, our analysis also suggests a few potential horizontal gene transfer events. Updated families and clans can be accessed through the new version of the FCPD database.
Conclusions
FCPD version 1.2 provides a systematic and searchable catalogue of 9,550 fungal CYP sequences (292 families) encoded by 108 fungal species and 147 CYP sequences (9 families) encoded by five oomycete species. In comparison to the first version, it offers a more comprehensive clan classification, is fully compatible with Nelson’s P450 databases, and has expanded functional categorization. These features will facilitate functional annotation and classification of CYPs encoded by newly sequenced fungal and oomycete genomes. Additionally, the classification system will aid in studying the roles of CYPs in the evolution of fungal adaptation to specific ecological niches.
Similar content being viewed by others

Background
Cytochrome P450 proteins (CYPs) are found in all domains of life[1] and represent one of the largest protein families. Their existence predates the emergence of oxygen-metabolizing life forms[2]. CYPs are defined by the absorption of light at 450nm by the heme cofactor, and oxidize a very diverse array of metabolic intermediates and environmental compounds. CYPs participate in a large number of primary, secondary and xenobiotic metabolic reactions[3].
The evolution of CYPs has been intimately intertwined with organismal adaptation to new ecological niches due to the roles of CYPs in the production of metabolites critical for specific processes such as pathogenesis, the utilization of specific substrates, and/or the detoxification of xenobiotics. Based on their roles in synthesizing or neutralizing toxic metabolites, many CYPs are hypothesized to have evolved through the chemical warfare waged among plants, animals, insects, and microbes[2, 4]. In fungi, several CYPs have been implicated in pathogen virulence because they neutralize antifungal compounds produced by hosts[5–7]. Expansions and diversifications of several CYP families have been associated with the evolution of fungal pathogenicity[8]. Accordingly, functional and evolutionary analyses of CYPs have been useful in understanding the ecological specialization and functional diversification of individual fungal taxa[9].
The extraordinary functional and evolutionary diversity of fungal CYPomes presents a major hurdle to CYP classification[10]. Fungal CYPs share little sequence similarity, except for a few conserved residues that are characteristic of CYPs. The most conserved region is the binding domain for a heme cofactor. Substrate binding regions are much more variable but may possess a signature motif. This motif is often found in conjunction with one or more binding domains such as those for cytochrome b5, ferredoxin, and binding sites for the NADPH cytochrome P450 reductase that contains FAD (flavin adenine dinucleotide) and FMN (flavin mononucleotide)[11].
Another challenge in develo** a comprehensive CYP classification system is the rapidly increasing number of sequenced fungal genomes. Currently, more than 250 genomes are present in the public domain[12, 13], but this number is predicted to increase rapidly (e.g.,http://1000.fungalgenomes.org). The rapid influx of genome sequences calls for robust computational tools that can effectively support large-scale comparative analyses of genomes and specific gene families.
The first nomenclature/grou** schema for CYPs, proposed by Nebert et al. in 1987[14], was based on amino acid sequence similarity. According to this schema, any two CYPs with sequence identity greater than 40% belong to a single CYP family; and any two CYPs with sequence identity greater than 55% belong to a subfamily. Manually curated databases of CYPs in multiple kingdoms based on this approach (thereafter referred to as Nelson’s P450 databases) have been maintained athttp://drnelson.uthsc.edu/CytochromeP450.html[15, 16]. These databases also serve as a central repository of CYP nomenclature. Unfortunately, this schema cannot be efficiently used to curate and classify rapidly increasing CYPs uncovered through genome sequencing.
The clan system approach was developed to support higher-level grou** of families identified via the sequence similarity-based schema. This approach places all CYP families with a monophyletic origin into a single clan and has been successfully applied to classify CYP families in Metazoa[17] and four fungal species[10]. For example, if new CYPs had equal identity to two or more CYP families, they can be tentatively assigned to a clan in which these families belong. Since the introduction of the “clan concept” in 1998 to classify metazoan CYPs[17], additional clans in vertebrates (9), plants (11)[18], arthropods[19], bivalves (4), and fungi (115)[10] have been identified. However, the clan classification system has become problematic for classifying the pan-fungal CYPome, because the number of fungal CYPs is too large to conduct phylogenetic analyses efficiently. Automated clustering based on sequence similarity remains the gold standard for the rapid classification of large protein sets[20, 21]. This approach does not require any prior knowledge and allows for rapid clustering of large protein families such as CYPs.
In 2008, we employed an automated clustering approach to build the Fungal Cytochrome P450 Database (FCPD)[22]. Since then the number of sequenced fungal genomes has increased substantially, which necessitated the improvement of our classification system. Additionally, the original FCPD classification generated several mega clusters, underscoring the need for optimizing clustering parameters.
Here we present FCPD release 1.2 (http://p450.riceblast.snu.ac.kr) with an improved CYP classification pipeline based on the modified TRIBE-MCL algorithm. The pipeline allowed for a larger number of CYP families to be merged into existing clans as well as supporting the discovery of potential new clans. To aid functional annotation, putative functional roles were assigned to over 150 clusters based on their similarity to functionally characterized fungal CYPs. The families and clans are accessible through FCPD, which offers global viewing and analysis of fungal CYPs.
Results and discussion
Identification of CYPs and optimization of clustering parameters
We first extracted all proteins that contained Interpro (http://www.ebi.ac.uk/interpro/) terms associated with CYPs from 324 genomes corresponding to 113 fungal and oomycete species, 94 other eukaryotic species, and six bacterial species (Figure1) as previously described[22]. While our main focus has been on curating fungal and oomycete CYPs, CYPs encoded by other eukaryotic species and selected bacterial species were included to aid in comparative evolutionary studies across kingdoms. Although oomycetes are fungus-like in that they produce hyphae and spores, they reside in a more basally derived eukaryotic lineage that includes chromophyte algae (Figure1). However, because mycologists have traditionally studied oomycetes, we analyzed CYPs from both true fungi and oomycetes. This data extraction resulted in 22,940 CYPs including 9,697 CYPs from fungi and oomycetes and 13,243 CYPs from other organisms (Figure1).

Phylogenetic relationships among taxa in FCPD 1.2 and the number of CYPs and clusters in each taxon. The tree topology based on Patterson and Sogin[23] is accompanied by a summary of the data archived in FCPD1.2. Each cluster may have CYPs from more than one phylum/subphylum. The number of clusters roughly corresponds to the number of CYP clans/families, thus illustrating the diversity of CYPs in each taxon.
Extracted protein sequences were clustered using an optimized protocol based on reciprocal pair-wise BLASTp all-against-all comparisons[24] followed by Tribe-MCL clustering[21] (see Methods and Additional file1 for details). The revision of the original clustering pipeline used to build FCPD[22] was motivated by a few factors, including the presence of many mega clusters with over 100 members, singlet clusters, and clusters that did not match families in Nelson’s P450 databases. While there are no absolute “best” criteria to optimize clustering, our main goal was to achieve more uniform grou** by minimizing the fractions of very large (>100 members) and singlet clusters.
Three parameters (E-value, inflation factor, and a new parameter called “coverage”) were evaluated and adjusted to optimize the performance [Additional file2]. Coverage was defined as the percentage of the query sequences matched by sequences from the database, thus the higher the coverage is, the lower is the possibility of false-positives. We tested patterns of clustering with various combinations of parameters in the optimum plane of a three-parameter space [Figure2] and settled on the following combination: E-value = 1e-50, inflation factor = 5, and coverage = 60%. The coverage parameter was instrumental in filtering out many false positives that display high E-values over short regions of similarity.

Optimizing parameters for clustering. Optimum values for the three parameters used for clustering with TribeMCL were chosen from the optimum plane consisting of the best possible combination of values (E-value, Inflation factor, and coverage).
CYP clustering in FCPD 1.2
Using the optimized parameters, we categorized 22,940 CYPs into 2,579 clusters (Figure1): fungal and oomycete CYPs belong to 1,090 (42%) clusters, while the remaining clusters (1,489) contained only non-fungal CYPs. Although there are a few clusters that contain CYPs from more than one kingdom, most clusters are kingdom-specific. All oomycete clusters consist of CYPs in oomycete species with the exception of one that also contains CYPs in plants, fungi, and protists. Among the non-fungal clusters, 778 clusters contained plant CYPs and 652 clusters contained metazoan CYPs.
To validate our clustering approach and to link resulting clusters to results from previous classifications, the clusters were compared with CYP families and clans identified in previous studies[10, 17], which in most instances showed good concordance between FCPD clusters and known families and clans. Out of 459 fungal CYP families identified in Nelson’s P450 databases, 292 matched with the CYPs in FCPD. Those that did not match corresponded to CYPs in species that are not currently covered in FCPD.
At the clan level, 77 clusters matched with 115 clans identified in a previous clan classification (Additional file3) with some clusters including multiple clans. In only three instances our clustering results suggested that two or more clans needed to be merged: (i) clans CYP531 and CYP532; (ii) CYP619 and CYP530; and (iii) CYP567, CYP561, CYP563, and CYP60. Orphan clans identified in the previous classification[10] were assigned to some of the non-orphan clans through our clustering. We identified 38 new putative clans and validated existing clans, which brought the total number of clans in FCPD to 117.
As a result of this expanded clan classification, 131 additional CYP families were put into new and existing clans (Additional file3). Of those, eight families that correspond to singlet FCPD clusters were classified as orphan clans. The resulting clans vary widely in size and number of CYP families included. The largest clans (CYP531 and CYP58) contain 14 families each. The size distribution analysis showed that, like many other protein families[25, 26], CYP clusters follow a power law distribution (Additional file4). Only 37 clusters with more than 100 members were observed. In contrast, 1,726 clusters were comprised of a single CYP. Information about individual clusters, families, and clans archived in FCPD will facilitate global analyses of fungal CYPs. New CYPs can be annotated using the BLAST search function.
Wide variation of the CYPome
The total number of CYPs and their relative fraction within the total proteome in different kingdoms and phyla varied widely. The boxplots in Figure3A show that plants have the largest CYPome (0.82%), bacteria have the smallest CYPome (0.05%), and fungi are placed in the middle (0.40%). The potato Solanum phureja has the largest CYPome composed of 629 CYPs.

Range of CYPome sizes across kingdoms and fungal phyla. The boxplot shows the average number of CYPs across (A) kingdoms and (B) fungal phyla.
The size of CYPome of individual species within kingdoms also varied drastically, presumably reflective of diverse lifestyles and ecologies. The largest variation was observed in fungi and plants. In fungi, Pezizomycotina and Basidiomycota have the largest and most variable CYPomes (Figure3B). The CYPome of certain basidiomycota fungi such as the brown rot fungus Postia placenta (353 CYPs) and the cocoa tree pathogen Moniliophthora perniciosa (307 CYPs) are larger than typical plant CYPomes. In these species, massive expansions of CYPs involved in oxidizing complex hydrocarbons were observed[27]. In contrast, some basidiomycota fungi, such as Puccinia graminis (18 CYPs) and Malassezia globosa (6 CYPs), have undergone massive reductions, probably reflecting their obligatory pathogenic lifestyles. Members of the Chytridiomycota and Oomycota also showed small CYPomes. Members of Saccharomycotina and Taphrinomycotina have the smallest CYPomes among fungi (2–3 CYPs).
Phyletic distribution of CYP families and clans in fungi
Our phyletic analysis showed an uneven distribution of CYP cluster sizes among taxa, which is consistent with extreme expansions and contractions of certain CYP families in the course of evolution. Seven out of the 30 largest fungal-specific clusters were exclusively composed of CYPs from the subphylum Pezizomycotina. The most dramatic expansions were observed in Pezizomycotina (clans CYP58 and CYP68), Agaricomycotina (clans CYP5150 and CYP63) and Mucoromycotina (family CYP509). Small clusters containing only species-specific CYPs were especially prevalent in members of Oomycota and Mucoromycotina.
The five largest fungal-specific clusters in FCPD had 1,056, 472, 452, 322, and 319 CYPs, respectively. These clusters represent some of the largest CYP families in fungi (Additional file5, Additional file6). The largest cluster (Cluster # 3) contains CYPs from the subphyla Agaricomycotina (Basidiomycota) and Pezizomycotina (Ascomycota). In this cluster, most Pezizomycotina CYPs (100) correspond to members of family CYP620, whereas 508 Agaricomycotina CYPs belong to family CYP5144. Some members of both families are known to be involved in xenobiotic metabolism[28]. Additionally, this cluster includes CYPs from the wood-rotting fungi Heterobasidion annosum (156) and Postia placenta (122), and more than 50 CYPs in six basidiomycete species, which suggests expansions of CYPs involved in the degradation of components of the wood (e.g., lignin, hemicellulose, cellulose).
The second largest fungal-specific cluster (# 11) has CYPs from Saccharomycotina and Pezizomycotina. It comprises the families CYP52, CYP548, CYP539, and CYP655 as well as a few other families involved in alkane assimilation (Additional file7). The third largest cluster (# 12) consists of CYPs from Pezizomycotina. The most dominant family in this cluster is CYP65, which contains CYPs predicted to function in secondary metabolism.
Six clusters contain both fungal and non-fungal CYPs, many of which are involved in evolutionary conserved core metabolic roles and are likely derived from common ancestral proteins. Cluster 17 contains family CYP61, one of the most conserved CYP families in fungi and beyond. The cluster has CYPs from all sub-phyla of fungi, Amoebozoa, and the unicellular diatom Capsaspora owczarzaki as well as one CYP from the algae Coccomyxa sp. Cluster 22 includes families CYP505 and CYP541, and CYPs from all fungal taxa, Actinobacteria, Bacillariophyta, and the plant Populus trichocarpa. Cluster 7 includes CYPs from Zygomycota and Blastocladiomycota as well as oomycetes, protists, and plants. Cluster 8 includes a single family from the chytrid Spizellomyces punctatus and many CYPs from chordates. Cluster 13 contains members of CYP51, which are implicated in sterol biosynthesis in all fungal phyla[29], and various CYPs from Amoebozoa, Bacillariophyta, Euglenozoa, and Chordata. Lastly, cluster 69 contains CYP55 family, in which fungal and bacterial CYPs are clustered together. Some of these families will be discussed in more detail below.
Our clustering approach also revealed 959 phylum-specific clusters and 1,044 CYPs that did not belong to any previously defined CYP families. Out of these, 560 were present in singlet clusters. CYP families present in individual phyla and subphyla (excluding Saccharomycotina) were also examined. Five CYP families were present in all species from Pezizomycotina and four families were present in all basidiomycete species, while 10 families were present in all species from Mucoromycotina. Among them, two families (CYP51 and CYP61) were common to all taxa. The CYP530 family is absent in the ascomycota fungi, however all the other basal lineages have retained this family (Figure4). The most parsimonious explanation is that CYP51, CYP61, and CYP530 were present in the last common ancestor of all fungi. Indeed, CYP51 is thought to be present even in early eukaryotes, and it has been hypothesized that CYP61 evolved from CYP51[30]. On the other hand, the family CYP530 seems to be specific to fungi and is known to be involved in degradation of various fatty acids and hydrocarbons (Additional file8: xenobiotic metabolism), allowing fungi to utilize these materials as nutrient sources.

Most conserved CYP families in fungi. CYP families were compared across different sub-phyla to determine the most conserved CYPs. CYP51 and CYP61 were present in all fungi except a couple of chytrids.
Functional annotation and classification of CYP clusters
To assign putative functional roles to individual clusters, we conducted a comprehensive literature review for functionally characterized fungal CYPs. This survey led to the identification of 54 CYPs that had been shown to be involved in (i) primary metabolism (15 CYPs), (ii) secondary metabolism (28) or (iii) xenobiotic metabolism (11) (Additional file5). We then used BLASTp to search the FCPD database with these CYPs as queries (Methods). A total of 2,457 hits (E-value cutoff of 1e-100) were generated with the CYPs involved in various primary metabolic reactions. This high number of hits is mainly due to the presence of well-conserved, house-kee** families such as CYP51 and CYP61, which are involved in ergosterol biosynthesis[29, 30]. Additionally, we found 544 and 642 hits with those CYPs involved in secondary and xenobiotic metabolism, respectively (Additional file9). Only one family (CYP58) contained CYPs involved in both secondary and xenobiotic metabolism. For instance, one such CYP58 gene from Phanerochaete chrysosporium has been predicted to function as benzoate 4-hydroxylase (xenobiotic metabolism) and at the same time is also involved in trichothecene biosynthesis (secondary metabolism)[31]. The relatively small number of hits to CYPs involved in secondary metabolism suggests that many fungi might have evolved a lineage-specific repertoire of CYPs to produce specific secondary metabolites.
Excluding CYP58, we found 12, 30, and 12 CYP families that uniquely matched to the primary, secondary, and xenobiotic metabolism categories, respectively. These 54 CYP families were then used to assign putative functional roles to the respective clans. With this approach we tentatively classified a total of 34 clans into primary (5 clans), secondary (17), and xenobiotic (12) metabolism (Additional file8).
Detailed analysis of specific clans
Selected CYP clans and families were analyzed in detail to augment and validate previous evolutionary studies[28–34] and to uncover notable features.
Clans 51 and 61
Our clustering analysis fully supported families CYP51 and CYP61, which are composed of house-kee** CYPs found in almost all fungi, plants and animals. CYP51 is a lanosterol 14-alpha demethylase involved in 14-demethylation of sterol precursors, and this demethylation step is common throughout all organisms[35]. To better understand its evolution, we constructed a phylogenetic tree with members of CYP51s from fungi, the early opisthokonts and other single-celled eukaryotes (Additional file10).
Most yeast species have a single CYP51 gene, whereas most Pezizomycotina species have two genes with the exception of Fusarium species and Aspergillus carbonarius (three genes). Basidiomycetes also have a single gene with the exception of Postia placenta and Coprinus cinereus (two genes). Rhizopus oryzae, Allomyces macrogynus, and Fragilariopsis cylindrus have two CYP51 genes and no CYP61 genes. This is consistent with the view that the CYP51 gene became duplicated very early in fungal evolution and then one of the duplicates may have given rise to CYP61[30].
CYP61 gene is a 22 sterol desaturase that carries out one of the last reactions in the Ergosterol metabolism pathway. The phylogenetic analysis of CYP61 (Additional file11) revealed the presence of a single gene in all yeasts and all basidiomycetes except P. placenta (two genes). Most ascomycota species have at least two genes with the exception of Pneumocystis carinii, as well as the basidiomycetes Puccinia graminis, and Melampsora laricis-populina, all three of which do not have a CYP61 gene. The absence of CYP61 genes in these species could be due to their obligate lifestyle, wherein they may utilize essential sterols from the plant/animal hosts.
Clans 65 and 68
Clans CYP65 and CYP68 consist of CYPs that belong to the secondary metabolism category. CYP65 has been found to catalyze the epoxidation reaction during the biosynthesis of the mycotoxin trichothecene, as well as during radicicol biosynthesis (Additional file5, Additional file12 and Additional file13). CYP68 carries out the C-8 oxygenation reaction during trichothecene biosynthesis (Additional file 5,Additional file 14) and the oxidation reaction during the biosynthesis of the plant hormone gibberellin[36]. The phylogenetic trees of CYP65 and CYP68 reveal multiple recent duplications and expansions (Additional file12, Additional file13 and Additional file14). These clans are absent in ascomycete yeasts and basidiomycete species, suggesting that they might have emerged in the ancestor of the Pezizomycotina.
Among members of the Pezizomycotina, there is a wide variation in the number of CYPs in clans CYP65 and CYP68. The Coccidioides species have just one CYP65 gene, whereas Dothideomycetes and Aspergillus species have 8–10 genes for CYP65s and 3–4 genes for CYP68s. Dothideomycetes have on average at least 5–6 more genes than other fungi, which is consistent with their secretion of diverse host-selective toxins (HSTs,[37]). Many of these HSTs are products of secondary metabolism pathways.
The highest number of CYP65 and CYP68 clan members is seen in Magnaporthe oryzae, Colletotrichum graminicola and Colletotrichum higginsianum (Additional file12 and Additional file14). All three fungi form appresoria (specialized infection structures formed by germinating spores) to enter the plant cell. Expression studies have demonstrated that secondary metabolism pathways are active during the infection process[38], suggesting that the increased number of CYP65 and CYP68 family members in these fungi might be linked to their pathogenicity.
Clan 505
CYP505 members are fatty acid hydroxylases that carry out the subterminal omega hydroxylation of fatty acids, a step required for the use of fatty acids as an energy source. It was hypothesized that CYP505 in fungi has evolved from the bacterial CYP450BM3 via a horizontal gene transfer (HGT) event[32]. This hypothesis is supported by the fact that both types have a fused NADPH CPR domain (http://drnelson.uthsc.edu/P4503d.html).
To test this HGT hypothesis, we performed a phylogenetic analysis of this clan (includes 161 CYPs from families CYP505 and CYP541). Contrary to the hypothesis, the tree topology (Additional file15) suggests an ancient origin of this clan in eukaryotes and subsequent losses in certain lineages. The earliest members of the clan seem to be present in the unicellular opisthokonts Capsaspora owczarzaki, Streptomyces species of bacteria and the unicellular algae Fragilariopsis cylindrus. There are at least two genes for CYP505 in most fungi, while early eukaryotes F. cylindrus and Allomyces macrogynus have 4–5 genes, suggesting an early increase in its copy number and subsequent gene losses. CYP505s are absent in ascomycete yeasts. Among members of the Pezizomycotina, A. flavus and Podospora anserina have five genes, and M. grisea has four genes. Basidiomycetes also have at least two genes with the white rot fungus P. chrysosporium containing six genes. It has been hypothesized[39] that CYP505 is used by plant-associated fungi to degrade plant cuticle which is synthesized by in-chain hydroxylation of fatty acids[40].
Clan 52
Cluster 11 contained all the CYPs belonging to clan CYP52. The highest numbers of CYP52 proteins (12) are seen in Aspergillus flavus, A. niger CBS 513.88, Trichoderma virens Gv29-8, Botrytis cinerea and Magnaporthe oryzae. Talaromyces stipitatus and Penicillium marneffei have 10 and 11 members of CYP52, respectively. In M. oryzae, CYP52 is upregulated during the penetration of the plant cuticle, which is made up of hydrocarbons[41]. Similar processes could be happening in B. cinerea and A. flavus, both of which are pathogenic to plants as well as Trichoderma virens Gv29-8 (twelve genes), T. reesei (nine genes), and T. atroviride (six genes) that are known to penetrate the fungal cell wall[42] as well as plant roots[43]. CYP52 genes are found in Candida species that are known to metabolize alkanes and other hydrocarbons, but are absent in Saccharomyces cerevisiae and Schizosaccharomyces pombe[44]. There were as many as 12 CYP52 proteins encoded by Yarrowia lipolytica, but there were no CYP52 proteins in basidiomycetes. All of these species might be using their CYP52 repertoire to support these processes, and expansion of the CYP52 family in these ascomycete fungi may allow efficient metabolism of various hydrocarbon compounds. We built a neighbor-joining tree to look at their evolutionary relationships (Additional file16). The most parsimonious evolutionary scenario suggests that the family evolved in the ancestor of budding yeasts but was lost in the lineage including S. cerevisiae but then expanded in the Pezizomycotina.
Clan 53 and Clan 504
CYP53 is a benzoate-para-hydroxylase enzyme that was first discovered in Aspergillus niger[45]. This benzoate detoxification occurs via the beta-ketoadipate pathway[46], which is present in many soil microbes that degrade aromatic compounds, some of which are released by plants[47]. Although benzoate detoxification appears to be the main function of members of this CYP group, some of them have also been found to exhibit O-demethylation activity[28]. Clan 53 is a single family clan in cluster 37 and contains 89 CYPs. This family is absent in ascomycete yeasts. A wide variation in its size was observed in the wood-decaying fungi Postia placenta (14 genes), Pleurotus osteratus (three genes) and Phanerochaete chrysosporium (one gene). Considering their proposed role in degrading plant-based aromatic compounds that are released by the plants into the soil or that might be present as a part of the dead plant material, this wide variation is puzzling. They are also present in several plant-pathogenic fungi such as Fusarium oxysporum (3), F. graminearum (4), Puccinia graminis (1), Moniliophthora perniciosa (2), Cochliobolus heterostrophus (3), and Botrytis cinerea (2), suggesting the possibility that the benzoate degrading activity may contribute to pathogenesis.
Clan CYP504 includes CYPs that are involved in phenylacetate catabolism[48]. Specifically, they are involved in the ortho-hydroxylation of phenylacetate, which is a precursor in penicillin production. Like Clan 53, this clan is a single-family cluster (cluster 29; Additional file17). The family is found in many saprophytic species as well as a number of basidiomycetes fungi that can degrade phenol derivatives as a source of carbon[49]. This family is also present in a number of human and plant-pathogenic fungi like Stagonospora nodorum (three genes), C. heterostrophus (four genes), Penicillium marneffei (five genes), Fusarium oxysporum (three genes), F. graminearum (four) and F. solani (five genes). Both CYP53 and CYP504 family members were found to be upregulated during cuticle infection by insect pathogenic fungi Metarhizium anisopliae (four genes) and M. acridum (two genes)[15]. To improve the classification, we added one more parameter, “coverage”, which was defined as the percentage of the query sequences matched by sequences from the database. To find optimal conditions for these three parameters, we tested efficiency of clustering with various combinations: (i) e-values between 1e-10 and 1e-100 at intervals of 1e-10; (ii) nine coverage values from 20% to 100% at intervals of 10%, and (iii) inflation factor from 1 to 5. We empirically chose optimal parameters as: e-value = 1e-50, coverage = 60%, and inflation factor = 5 (Additional file2).
Clan identification
We were able to expand the clans identified in earlier studies[10, 31, 69] through our optimized clustering procedure. We searched for each clan through our database using a search function that was built to facilitate searching the database using various terms (e.g., Sequence ID, taxonomic group, and CYP family). We followed this step for all the clans mentioned in previous studies[10, 17, 31, 69], which allowed us to identify novel clans and assign CYP families to previously identified orphan clans (Additional file3). There were a number of CYPs that did not show any significant similarity to any of the CYP families in Nelson’s P450 databases, indicating that they are members of novel CYP families. Most of them were present in singlet clusters.
Classification of CYPs into putative functional categories
An extensive literature search was performed to identify 54 functionally characterized fungal CYPs. These CYPs were then matched to CYPs in FCPD using BLASTp with an E-value cutoff of 1e-100. This stringent E-value was chosen based on an empirical testing of several E-values. Based on similarity to the characterized CYPs, CYP families were classified into three broad functional categories: (i) primary metabolism, (ii) secondary metabolism, and (iii) xenobiotic metabolism. Many of the hits occurred in more than one category. In order to link CYP clans into these functional categories, we have transferred functional annotations described above into respective clans. The BLASTp hits and the characterized set of CYPs can be accessed athttp://p450.riceblast.snu.ac.kr/char_p450.php.
Online database architecture
FCPD has been developed using PHP script with MySQL database[22]. The Linux-based apache web-server and task management system supports BLAST analysis and MCL clustering. The middle-ware written in Perl script simultaneously executes the bioinformatics pipelines from the query submitted by the end-user, and retrieves the archived CYP dataset. The pipeline for FCPD can be found in Additional file1.
References
Bernhardt R: Cytochromes P450 as versatile biocatalysts. J Biotechnol. 2006, 124 (1): 128-145. 10.1016/j.jbiotec.2006.01.026.
Lewis DF, Watson E, Lake BG: Evolution of the cytochrome P450 superfamily: sequence alignments and pharmacogenetics. Mutat Res. 1998, 410 (3): 245-270. 10.1016/S1383-5742(97)00040-9.
Guengerich FP: Cytochrome p450 and chemical toxicology. Chem Res Toxicol. 2008, 21 (1): 70-83. 10.1021/tx700079z.
Gonzalez FJ, Nebert DW: Evolution of the P450 gene superfamily: animal-plant ’warfare’, molecular drive and human genetic differences in drug oxidation. Trends Genet. 1990, 6 (6): 182-186.
Leal GA, Gomes LH, Albuquerque PS, Tavares FC, Figueira A: Searching for Moniliophthora perniciosa pathogenicity genes. Fungal Biol. 2010, 114 (10): 842-854. 10.1016/j.funbio.2010.07.009.
Maloney AP, VanEtten HD: A gene from the fungal plant pathogen Nectria haematococca that encodes the phytoalexin-detoxifying enzyme pisatin demethylase defines a new cytochrome P450 family. Mol Gen Genet. 1994, 243 (5): 506-514. 10.1007/BF00284198.
Siewers V, Viaud M, Jimenez-Teja D, Collado IG, Gronover CS, Pradier JM, Tudzynski B, Tudzynski P: Functional analysis of the cytochrome P450 monooxygenase gene bcbot1 of Botrytis cinerea indicates that botrydial is a strain-specific virulence factor. Mol Plant Microbe Interact. 2005, 18 (6): 602-612. 10.1094/MPMI-18-0602.
Soanes DM, Alam I, Cornell M, Wong HM, Hedeler C, Paton NW, Rattray M, Hubbard SJ, Oliver SG, Talbot NJ: Comparative genome analysis of filamentous fungi reveals gene family expansions associated with fungal pathogenesis. PLoS One. 2008, 3 (6): e2300-10.1371/journal.pone.0002300.
Soanes DM, Richards TA, Talbot NJ: Insights from sequencing fungal and oomycete genomes: what can we learn about plant disease and the evolution of pathogenicity?. Plant Cell. 2007, 19 (11): 3318-3326. 10.1105/tpc.107.056663.
Deng J, Carbone I, Dean RA: The evolutionary history of cytochrome P450 genes in four filamentous Ascomycetes. BMC Evol Biol. 2007, 7: 30-10.1186/1471-2148-7-30.
Cha CJ, Doerge DR, Cerniglia CE: Biotransformation of malachite green by the fungus Cunninghamella elegans. Appl Environ Microbiol. 2001, 67 (9): 4358-4360. 10.1128/AEM.67.9.4358-4360.2001.
Park J, Park B, Jung K, Jang S, Yu K, Choi J, Kong S, Kim S, Kim H, Kim JF, et al: CFGP: a web-based, comparative fungal genomics platform. Nucleic Acids Res. 2008, 36: D562-D571-
Jung K, Park J, Choi J, Park B, Kim S, Ahn K, Choi D, Kang S, Lee YH: SNUGB: a versatile genome browser supporting comparative and functional fungal genomics. BMC Genomics. 2008, 9: 586-10.1186/1471-2164-9-586.
Nebert DW, Adesnik M, Coon MJ, Estabrook RW, Gonzalez FJ, Guengerich FP, Gunsalus IC, Johnson EF, Kemper B, Levin W, et al: The P450 gene superfamily: recommended nomenclature. DNA. 1987, 6 (1): 1-11. 10.1089/dna.1987.6.1.
Nelson DR, Kamataki T, Waxman DJ, Guengerich FP, Estabrook RW, Feyereisen R, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, et al: The P450 superfamily: update on new sequences, gene map**, accession numbers, early trivial names of enzymes, and nomenclature. DNA Cell Biol. 1993, 12 (1): 1-51. 10.1089/dna.1993.12.1.
Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, et al: P450 superfamily: update on new sequences, gene map**, accession numbers and nomenclature. Pharmacogenetics. 1996, 6 (1): 1-42. 10.1097/00008571-199602000-00002.
Nelson DR: Metazoan cytochrome P450 evolution. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 1998, 121 (1–3): 15-22.
Nelson D, Werck-Reichhart D: A P450-centric view of plant evolution. Plant J. 2011, 66 (1): 194-211. 10.1111/j.1365-313X.2011.04529.x.
Feyereisen R: Arthropod CYPomes illustrate the tempo and mode in P450 evolution. Biochim Biophys Acta. 2011, 1814 (1): 19-28. 10.1016/j.bbapap.2010.06.012.
Krause A, Stoye J, Vingron M: Large scale hierarchical clustering of protein sequences. BMC Bioinforma. 2005, 6: 15-10.1186/1471-2105-6-15.
Enright AJ, Van Dongen S, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30 (7): 1575-1584. 10.1093/nar/30.7.1575.
Park J, Lee S, Choi J, Ahn K, Park B, Kang S, Lee YH: Fungal cytochrome P450 database. BMC Genomics. 2008, 9: 402-10.1186/1471-2164-9-402.
Medina EM, Jones GW, Fitzpatrick DA: Reconstructing the fungal tree of life using phylogenomics and a preliminary investigation of the distribution of yeast prion-like proteins in the fungal kingdom. J Mol Evol. 2011, 73 (3–4): 116-133.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215 (3): 403-410.
Reed WJ, Hughes BD: A model explaining the size distribution of gene and protein families. Math Biosci. 2004, 189 (1): 97-102. 10.1016/j.mbs.2003.11.002.
Unger R, Uliel S, Havlin S: Scaling law in sizes of protein sequence families: from super-families to orphan genes. Proteins. 2003, 51 (4): 569-576. 10.1002/prot.10347.
Syed K, Doddapaneni H, Subramanian V, Lam YW, Yadav JS: Genome-to-function characterization of novel fungal P450 monooxygenases oxidizing polycyclic aromatic hydrocarbons (PAHs). Biochem Biophys Res Commun. 2010, 399 (4): 492-497. 10.1016/j.bbrc.2010.07.094.
Ide M, Ichinose H, Wariishi H: Molecular identification and functional characterization of cytochrome P450 monooxygenases from the brown-rot basidiomycete Postia placenta. Arch Microbiol. 2012, 194 (4): 243-253. 10.1007/s00203-011-0753-2.
Aoyama Y, Noshiro M, Gotoh O, Imaoka S, Funae Y, Kurosawa N, Horiuchi T, Yoshida Y: Sterol 14-demethylase P450 (P45014DM*) is one of the most ancient and conserved P450 species. J Biochem. 1996, 119 (5): 926-933. 10.1093/oxfordjournals.jbchem.a021331.
Kelly SL, Lamb DC, Baldwin BC, Corran AJ, Kelly DE: Characterization of Saccharomyces cerevisiae CYP61, sterol delta22-desaturase, and inhibition by azole antifungal agents. J Biol Chem. 1997, 272 (15): 9986-9988. 10.1074/jbc.272.15.9986.
Doddapaneni H, Chakraborty R, Yadav JS: Genome-wide structural and evolutionary analysis of the P450 monooxygenase genes (P450ome) in the white rot fungus Phanerochaete chrysosporium: evidence for gene duplications and extensive gene clustering. BMC Genomics. 2005, 6: 92-10.1186/1471-2164-6-92.
Kitazume T, Takaya N, Nakayama N, Shoun H: Fusarium oxysporum fatty-acid subterminal hydroxylase (CYP505) is a membrane-bound eukaryotic counterpart of Bacillus megaterium cytochrome P450BM3. J Biol Chem. 2000, 275 (50): 39734-39740. 10.1074/jbc.M005617200.
Zimmer T, Ohkuma M, Ohta A, Takagi M, Schunck WH: The CYP52 multigene family of Candida maltosa encodes functionally diverse n-alkane-inducible cytochromes P450. Biochem Biophys Res Commun. 1996, 224 (3): 784-789. 10.1006/bbrc.1996.1100.
Craft DL, Madduri KM, Eshoo M, Wilson CR: Identification and characterization of the CYP52 family of Candida tropicalis ATCC 20336, important for the conversion of fatty acids and alkanes to alpha, omega-dicarboxylic acids. Appl Environ Microbiol. 2003, 69 (10): 5983-5991. 10.1128/AEM.69.10.5983-5991.2003.
Aoyama Y: Recent progress in the CYP51 research focusing on its unique evolutionary and functional characteristics as a diversozyme P450. Front Biosci. 2005, 10: 1546-1557. 10.2741/1639.
Tudzynski B: Gibberellin biosynthesis in fungi: genes, enzymes, evolution, and impact on biotechnology. Appl Microbiol Biotechnol. 2005, 66 (6): 597-611. 10.1007/s00253-004-1805-1.
Ciuffetti LM, Manning VA, Pandelova I, Betts MF, Martinez JP: Host-selective toxins, Ptr ToxA and Ptr ToxB, as necrotrophic effectors in the Pyrenophora tritici-repentis-wheat interaction. New Phytol. 2010, 187 (4): 911-919. 10.1111/j.1469-8137.2010.03362.x.
Collemare J, Pianfetti M, Houlle AE, Morin D, Camborde L, Gagey MJ, Barbisan C, Fudal I, Lebrun MH, Bohnert HU: Magnaporthe grisea avirulence gene ACE1 belongs to an infection-specific gene cluster involved in secondary metabolism. New Phytol. 2008, 179 (1): 196-208. 10.1111/j.1469-8137.2008.02459.x.
Deng J: Structural, Functional and Evolutionary Analyses of the Rice Blast Fungal Genome. 2006, North Carolina State University, Raleigh
Salaun JP, Helvig C: Cytochrome P450-dependent oxidation of fatty acids. Drug Metabol Drug Interact. 1995, 12 (3–4): 261-283.
Oh Y, Donofrio N, Pan H, Coughlan S, Brown DE, Meng S, Mitchell T, Dean RA: Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus Magnaporthe oryzae. Genome Biol. 2008, 9 (5): R85-10.1186/gb-2008-9-5-r85.
Gruber S, Seidl-Seiboth V: Self vs. non-self: Fungal cell wall degradation in Trichoderma. Microbiology. 2012, 158 (Pt 1): 26-34.
Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK, et al: Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011, 12 (4): R40-10.1186/gb-2011-12-4-r40.
Ohkuma M, Muraoka S, Tanimoto T, Fujii M, Ohta A, Takagi M: CYP52 (cytochrome P450alk) multigene family in Candida maltosa: identification and characterization of eight members. DNA Cell Biol. 1995, 14 (2): 163-173. 10.1089/dna.1995.14.163.
van Gorcom RF, Boschloo JG, Kuijvenhoven A, Lange J, van Vark AJ, Bos CJ, van Balken JA, Pouwels PH, van den Hondel CA: Isolation and molecular characterisation of the benzoate-para-hydroxylase gene (bphA) of Aspergillus niger: a member of a new gene family of the cytochrome P450 superfamily. Mol Gen Genet. 1990, 223 (2): 192-197.
Harwood CS, Parales RE: The beta-ketoadipate pathway and the biology of self-identity. Annu Rev Microbiol. 1996, 50: 553-590. 10.1146/annurev.micro.50.1.553.
Bénigne-Ernest Amborabéa PF-L, Jean-François C, Gabriel R: Antifungal effects of salicylic acid and other benzoic acid derivatives towards Eutypa lata: structure–activity relationship. Plant Physiol Biochem. 2003, 40 (12): 1051-1060.
Mingot JM, Penalva MA, Fernandez-Canon JM: Disruption of phacA, an Aspergillus nidulans gene encoding a novel cytochrome P450 monooxygenase catalyzing phenylacetate 2-hydroxylation, results in penicillin overproduction. J Biol Chem. 1999, 274 (21): 14545-14550. 10.1074/jbc.274.21.14545.
Lah L, Podobnik B, Novak M, Korosec B, Berne S, Vogelsang M, Krasevec N, Zupanec N, Stojan J, Bohlmann J, et al: The versatility of the fungal cytochrome P450 monooxygenase system is instrumental in xenobiotic detoxification. Mol Microbiol. 2011, 81 (5): 1374-1389. 10.1111/j.1365-2958.2011.07772.x.
Gao Q, ** K, Ying SH, Zhang Y, **ao G, Shang Y, Duan Z, Hu X, **e XQ, Zhou G, et al: Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011, 7 (1): e1001264-10.1371/journal.pgen.1001264.
Mendonca Ade L, da Silva CE, de Mesquita FL, Campos Rda S, Do Nascimento RR, **menes EC, Sant’Ana AE: Antimicrobial activities of components of the glandular secretions of leaf cutting ants of the genus Atta. Antonie Van Leeuwenhoek. 2009, 95 (4): 295-303. 10.1007/s10482-009-9312-0.
Tyler BM, Tripathy S, Zhang X, Dehal P, Jiang RH, Aerts A, Arredondo FD, Baxter L, Bensasson D, Beynon JL, et al: Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006, 313 (5791): 1261-1266. 10.1126/science.1128796.
Goddard MR, Burt A: Recurrent invasion and extinction of a selfish gene. Proc Natl Acad Sci USA. 1999, 96 (24): 13880-13885. 10.1073/pnas.96.24.13880.
Holst-Jensen A, Vaage M, Schumacher T, Johansen S: Structural characteristics and possible horizontal transfer of group I introns between closely related plant pathogenic fungi. Mol Biol Evol. 1999, 16 (1): 114-126. 10.1093/oxfordjournals.molbev.a026031.
Collins RA, Saville BJ: Independent transfer of mitochondrial chromosomes and plasmids during unstable vegetative fusion in Neurospora. Nature. 1990, 345 (6271): 177-179. 10.1038/345177a0.
Khaldi N, Wolfe KH: Elusive origins of the extra genes in Aspergillus oryzae. PLoS One. 2008, 3 (8): e3036-10.1371/journal.pone.0003036.
Lawrence JG, Roth JR: Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics. 1996, 143 (4): 1843-1860.
Campbell MA, Rokas A, Slot JC: Horizontal transfer and death of a fungal secondary metabolic gene cluster. Genome Biol Evol. 2012, 4 (3): 289-293. 10.1093/gbe/evs011.
Yadav JS, Doddapaneni H, Subramanian V: P450ome of the white rot fungus Phanerochaete chrysosporium: structure, evolution and regulation of expression of genomic P450 clusters. Biochem Soc Trans. 2006, 34 (Pt 6): 1165-1169.
Lodeiro S, **ong Q, Wilson WK, Ivanova Y, Smith ML, May GS, Matsuda SP: Protostadienol biosynthesis and metabolism in the pathogenic fungus Aspergillus fumigatus. Org Lett. 2009, 11 (6): 1241-1244. 10.1021/ol802696a.
Mitsuguchi H, Seshime Y, Fujii I, Shibuya M, Ebizuka Y, Kushiro T: Biosynthesis of steroidal antibiotic fusidanes: functional analysis of oxidosqualene cyclase and subsequent tailoring enzymes from Aspergillus fumigatus. J Am Chem Soc. 2009, 131 (18): 6402-6411. 10.1021/ja8095976.
Rehman S, Shawl AS, Verma V, Kour A, Athar M, Andrabi R, Sultan P, Qazi GN: An endophytic Neurospora sp. from Nothapodytes foetida producing camptothecin. Prikl Biokhim Mikrobiol. 2008, 44 (2): 225-231.
Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, Wasmann CC, Grimwood J, Schmutz J, Taga M, White GJ, Zhou S, et al: The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009, 5 (8): e1000618-10.1371/journal.pgen.1000618.
Temporini ED, VanEtten HD: An analysis of the phylogenetic distribution of the pea pathogenicity genes of Nectria haematococca MPVI supports the hypothesis of their origin by horizontal transfer and uncovers a potentially new pathogen of garden pea: Neocosmospora boniensis. Curr Genet. 2004, 46 (1): 29-36.
Pinot F, Beisson F: Cytochrome P450 metabolizing fatty acids in plants: characterization and physiological roles. FEBS J. 2011, 278 (2): 195-205. 10.1111/j.1742-4658.2010.07948.x.
Roberts GA, Celik A, Hunter DJ, Ost TW, White JH, Chapman SK, Turner NJ, Flitsch SL: A self-sufficient cytochrome p450 with a primary structural organization that includes a flavin domain and a [2Fe-2S] redox center. J Biol Chem. 2003, 278 (49): 48914-48920. 10.1074/jbc.M309630200.
Hunter DJ, Roberts GA, Ost TW, White JH, Muller S, Turner NJ, Flitsch SL, Chapman SK: Analysis of the domain properties of the novel cytochrome P450 RhF. FEBS Lett. 2005, 579 (10): 2215-2220. 10.1016/j.febslet.2005.03.016.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S: MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011, 28 (10): 2731-2739. 10.1093/molbev/msr121.
Petric S, Hakki T, Bernhardt R, Zigon D, Cresnar B: Discovery of a steroid 11alpha-hydroxylase from Rhizopus oryzae and its biotechnological application. J Biotechnol. 2010, 150 (3): 428-437. 10.1016/j.jbiotec.2010.09.928.
Hartman H, Matsuno K: The Origin and Evolution of the Cell: Proceedings of the Conference on the Origin and Evolution of Prokaryotic and Eukaryotic Cells: 22-25 April 1992. 1993, World Scientific Pub Co Inc, Shimoda, Japan, Singapore
Acknowledgements
This research has been supported by the USDA Agriculture and Food Research Initiative Competitive Grants Program (Grant no. 2010-65110-20488). The work in Lee’s lab has been supported by the National Research Foundation of Korea (2012–0001149 and 2012–0000141) and the Next-Generation Bio-Green 21 Program of Rural Development Administration in Korea (PJ00821201). The authors would like to thank Douglas Whalen for lending his voice for the FCPD 1.2 video tutorials and for reviewing the paper and Jill Demers for reviewing the paper.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
VPM wrote the draft manuscript, designed the pipeline and carried out all the detailed analyses. JP set up the database and designed the pipeline. BP and JC contributed to database improvement and data curation. NF-A co-wrote the manuscript. SK and Y-HL conceptualized and coordinated the study and guided manuscript preparation. All authors read and approved the final manuscript.
Electronic supplementary material
12864_2012_4388_MOESM1_ESM.pdf
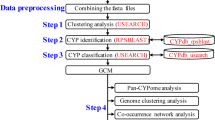
Additional file 1: Pipeline employed in FCPD 1.2 version. The pipeline still consists of four steps employed in building the previous version of FCPD, but step 3 is now based on optimized parameters. Additionally, a new parameter, coverage, was added to the clustering procedure to further improve clustering results. (PDF 235 kb) (PDF 235 KB)
12864_2012_4388_MOESM4_ESM.pdf
Additional file 4: CYP family sizes follow a power law distribution. The graph shows the family size distribution across families. (PDF 167 kb) (PDF 167 KB)
12864_2012_4388_MOESM12_ESM.tiff
Additional file 12: Phylogenetic tree of CYP65 in Pezizomycotina. Basidiomycetes and Ascomycete yeast species lack family CYP65. In Pezizomycotina, there is large variation in the number of CYP65 family genes. Coccidioides and Neurospora spp. have only one and two members, respectively. On the other hand, Dothideomycetes fungi have 6–15 members. The tree was adapted from Medina et al.[70]. (TIFF 7901 kb) (TIFF 8 MB)
12864_2012_4388_MOESM14_ESM.tiff
Additional file 14: Phylogenetic tree of CYP68. CYP68 family members are found in a number of secondary metabolism gene clusters. This family was lost in yeasts, and is absent in most Basidiomycetes except for some Homobasidiomycetes species. The tree was adapted from Medina et al.[70]. (TIFF 8118 kb) (TIFF 8 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Moktali, V., Park, J., Fedorova-Abrams, N.D. et al. Systematic and searchable classification of cytochrome P450 proteins encoded by fungal and oomycete genomes. BMC Genomics 13, 525 (2012). https://doi.org/10.1186/1471-2164-13-525
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2164-13-525