
Abstract
Serum response factor (SRF) regulates certain microRNAs that play a role in cardiac and skeletal muscle development. However, the role of SRF in the regulation of microRNA expression and microRNA biogenesis in cardiac hypertrophy has not been well established. In this report, we employed two distinct transgenic mouse models to study the impact of SRF on cardiac microRNA expression and microRNA biogenesis. Cardiac-specific overexpression of SRF (SRF-Tg) led to altered expression of a number of microRNAs. Interestingly, downregulation of miR-1, miR-133a and upregulation of miR-21 occurred by 7 days of age in these mice, long before the onset of cardiac hypertrophy, suggesting that SRF overexpression impacted the expression of microRNAs which contribute to cardiac hypertrophy. Reducing cardiac SRF level using the antisense-SRF transgenic approach (Anti-SRF-Tg) resulted in the expression of miR-1, miR-133a and miR-21 in the opposite direction. Furthermore, we observed that SRF regulates microRNA biogenesis, specifically the transcription of pri-microRNA, thereby affecting the mature microRNA level. The mir-21 promoter sequence is conserved among mouse, rat and human; one SRF binding site was found to be in the mir-21 proximal promoter region of all three species. The mir-21 gene is regulated by SRF and its cofactors, including myocardin and p49/Strap. Our study demonstrates that the downregulation of miR-1, miR-133a, and upregulation of miR-21 can be reversed by one single upstream regulator, SRF. These results may help to develop novel therapeutic interventions targeting microRNA biogenesis.
Similar content being viewed by others
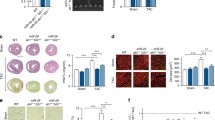

Background
MicroRNAs (miRNAs) are short (20 to 23-nucleotide), endogenous, single-stranded RNA molecules that regulate gene expression by hybridization to messenger RNAs (mRNAs) with the consequence of mRNA degradation or translational inhibition of targeted transcripts. Genes that encode for miRNA are transcribed by either RNA polymerase II or RNA polymerase III into primary miRNA (pri-miRNA) transcripts, which are then cleaved by the nuclear microprocessor complex formed by the RNase III enzyme Drosha (RNASEN) and the DGCR8 (DiGeorge critical region 8) protein. The RNase III Dicer cleaves off the loop of the pre-miRNA to generate a roughly 22-nucleotide miRNA duplex [1].
Although insights into the regulatory function of miRNAs toward their mRNA targets are beginning to emerge, less is known about the regulation of miRNA gene expression and miRNA biogenesis [1]. For instance, it has been shown that miRNAs participate in the control of cardiac development, and a number of miRNAs play a role in cardiac hypertrophy [2–5]. In addition, serum response factor (SRF), an important transcription factor, participates in the regulation of several cardiac enriched miRNAs, including mir-1 and mir-133a [4, 6]. However, it is unclear at what specific stage SRF regulates the biogenesis of miRNA.
SRF is a member of the MADS (MCM1, agamous, deficiens, SRF) family of transcriptional activators that has been implicated in the regulation of a number of genes that are important in cell proliferation and differentiation. SRF regulates its target genes by binding to the serum response element (SRE), which contains a consensus CC(A/T6)GG (CArG) motif [7–9]. This cognate response site of SRF is found in the promoter region of certain immediate-early genes and many muscle-specific genes [9–30]. The miRNA profile has been shown to serve as an impressive phenotypic signature. Therefore, miRNA has the potential to be developed as diagnostic and/or prognostic tools [31]. On the other hand, since individual miRNAs are dysregulated either negatively or positively with disease, strategies are needed to counteract or reverse the altered expression, potentially through reconstituting and/or targeting regimens, respectively [32–34]. A deeper understanding of miRNA biogenesis, as well as the regulation of each step in the miRNA maturation process and the involvement of signaling pathways and transcription factors is needed.
It has been reported that miRNAs undergo multiple steps during biogenesis, in which three forms of miRNAs are produced: pri-miRNA, pre-miRNA and mature miRNA [1, 35]. Multiple proteins are involved in this process. RNA polymerase II or RNA polymerase III transcribes the transcripts of pri-miRNA; a microprocessor complex cleaves pri-miRNA to generate pre-miRNA; and another endonuclease, Dicer, cleaves the pre-miRNA to release a miRNA-miRNA duplex. The miRNA strand is then stripped away from the duplex to leave a mature, approximately 22-nucleotide miRNA [36]. These individual steps can be targeted to increase or decreaase the miRNA level. Our data reveal that SRF executes it regulatory role in miRNA biogenesis through transcriptional regulation where it controls the amount of pri-miRNA available for downstream miRNA maturation process. We have proposed a model of miRNA regulation by SRF and its cofactors (Figure 10).

A model of miRNA regulation mediated by SRF and its cofactors, including myocardin, p49/Strap, Zipzap and other proteins. Generally, the pri-miRNA transcript contains one miRNA (e.g pri-mir-21), but it can also contain more than one miRNAs (e.g. mir-1 and mir-133a). SRF regulates the transcription activity of both types of pri-miRNAs, thereby affecting the downstream mature miRNA level. The length of three representative primary transcripts: pri-mir-21 is over 3 kb, "pri-mir-1-1 and pri-mir-133a2" is 10 kb, and "pri-mir-1-2 and pri-mir-133a1" is 6 kb.
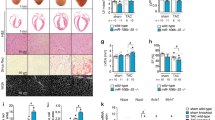
Mir-21 is known to be up-regulated in many forms of cancer as well as in the heart during cardiac hypertrophic growth and heart failure [37–40]. In the failing heart, miR-21 levels are increased in fibroblasts where it inhibits sprouty homologue 1 (Spry1) and promotes fibrosis [16]. In vivo, the silencing of miR-21 by a specific "antagomir" in a mouse pressure-overload-induced disease model was reported to reduce cardiac ERK-MAP kinase activity, inhibit interstitial fibrosis and attenuate cardiac dysfunction [16]. It has been reported that TGF-β and BMP signaling promote a rapid increase in the expression of mature miR-21 at the post-transcriptional level through accelerating the process of pri-miR-21 into precursor miR-21 (pre-miR-21) by the DROSHA complex [41]. Mir-21 is one of the miRNAs that was significantly increased in the SRF-Tg heart, but was decreased in the anti-SRF-Tg heart. However, the mir-21 gene promoter was repressed by SRF and p49/Strap in transfection assay, and the promoter was strongly induced by myocardin. Similarly, we observed that SRF activates both cardiac α-actin and MLC-2v promoter-luciferase DNA constructs in vitro assay, but SRF repressed the expression of both mRNA in the mouse heart [18, 22]. These data indicate that the regulation of mir-21 gene expression is complex, and involves SRF in coordination with multiple SRF cofactors, including myocardin, p49/Strap and Zipzap. The fact that a "knock-down" of SRF level could decrease the mir-21 level provides an opportunity for future studies manipulating mir-21 level through the SRF-mediated signaling pathway.
Both miR-1 and miR-133a are produced from the same polycistronic transcripts, which are encoded by two separate genes in the mouse and the human genomes [42]. The mouse pri-mir-1-1 and pri-mir-133a-2 are transcribed into a polycistronic transcript that is 10 kb in length, and the pri-mir-1-2 and pri-mir-133a-1 are transcribed into another polycistronic transcript that is 6 kb in length [42]. Our data revealed that the down-regulation of miR-1 correlates closely with that of miR-133a in the SRF-Tg at various time points from 7 days to 6 months of age (Figure 7B). These findings suggest that SRF may regulate these two miRNAs at the level of polycistronic transcription, rather than at each individual miRNA (pri-mir-1 or pri-mir-133a) transcription, thereby kee** the expression of both miRNAs closely correlated. Since mir-1-1 and mir-1-2 genes are located on two different chromosomes, their expression is divergent. The pri-mir-1-1 is expressed at 6-fold higher than pri-mir-1-2. Therefore, the contribution of pri-mir-1-1 to the mature miR-1 pool may be greater than that of pri-mir-1-2. Given the fact that targeted mutation of mir-1-2 gene resulted in embryonic myocardial dysfunction and half of the mutant mice suffered early death due to ventricular septal defect (VSD) [4], one might speculate that a targeted mutation of mir-1-1 gene would also cause equally (or more) severe consequences.
The miR-1 is the most abundant miRNA that is expressed in the heart. Our present study revealed that miR-1 accounted for 7% of all the 578 miRNAs detected by the microarray. Mir-1 and mir-133a are down-regulated in cardiac hypertrophy and cardiac failure, suggesting that they may play a role in the underlying pathogenesis [14, 43]. It is plausible that increasing mir-1 and mir-133a level at a specific time point may have potentially beneficial effects against the pathological conditions. Matkovich et al reported that an increase of mir-133a level in the postnatal heart has beneficial effects against cardiac fibrosis after transverse aortic constriction [44].
In conclusion, our current study demonstrates that cardiac-specific overexpression of SRF leads to altered expression of cardiac miRNAs, especially the down-regulation of miR-1 and miR-133a, and up-regulation of miR-21, the dysregulation of which is known to contribute to cardiac hypertrophy. We observed that SRF plays a role in the regulation of miRNA biogenesis, specifically at the level of transcription of pri-miRNA. Reducing cardiac SRF level using the antisense-SRF transgenic approach led to the expression of miR-1, miR-133a and miR-21 in the opposite direction to that of SRF overexpression. Our findings may help in the future development of therapeutic interventions through targeting of the SRF-mediated signaling pathways.
References
Winter J, Jung S, Keller S, Gregory RI, Diederichs S: Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009, 11 (3): 228-234. 10.1038/ncb0309-228.
Kwon C, Han Z, Olson EN, Srivastava D: MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proc Natl Acad Sci USA. 2005, 102 (52): 18986-18991. 10.1073/pnas.0509535102.
van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN: A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci USA. 2006, 103 (48): 18255-18260. 10.1073/pnas.0608791103.
Zhao Y, Samal E, Srivastava D: Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005, 436 (7048): 214-220. 10.1038/nature03817.
Callis TE, Chen JF, Wang DZ: MicroRNAs in skeletal and cardiac muscle development. DNA Cell Biol. 2007, 26 (4): 219-225. 10.1089/dna.2006.0556.
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN: microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22 (23): 3242-3254. 10.1101/gad.1738708.
Treisman R: Identification of a protein-binding site that mediates transcriptional response of the c-fos gene to serum factors. Cell. 1986, 46 (4): 567-574. 10.1016/0092-8674(86)90882-2.
Boxer LM, Prywes R, Roeder RG, Kedes L: The sarcomeric actin CArG-binding factor is indistinguishable from the c-fos serum response factor. Mol Cell Biol. 1989, 9 (2): 515-522.
Miano JM: Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003, 35 (6): 577-593. 10.1016/S0022-2828(03)00110-X.
Zilberman A, Dave V, Miano J, Olson EN, Periasamy M: Evolutionarily conserved promoter region containing CArG*-like elements is crucial for smooth muscle myosin heavy chain gene expression. Circ Res. 1998, 82 (5): 566-575.
Nelson TJ, Balza R, **ao Q, Misra RP: SRF-dependent gene expression in isolated cardiomyocytes: regulation of genes involved in cardiac hypertrophy. J Mol Cell Cardiol. 2005, 39 (3): 479-489. 10.1016/j.yjmcc.2005.05.004.
Zhang X, Azhar G, Furr MC, Zhong Y, Wei JY: Model of functional cardiac aging: young adult mice with mild overexpression of serum response factor. Am J Physiol Regul Integr Comp Physiol. 2003, 285 (3): R552-560.
Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M: MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007, 100 (3): 416-424. 10.1161/01.RES.0000257913.42552.23.
Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G: MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007, 13 (5): 613-618. 10.1038/nm1582.
Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, Rojas M, Hammond SM, Wang DZ: Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007, 42 (6): 1137-1141. 10.1016/j.yjmcc.2007.04.004.
Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S: MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008, 456 (7224): 980-984. 10.1038/nature07511.
Azhar G, Zhang X, Zuo C, Wang CS, Wei JY: Mildly reduced SRF protein is good for the older heart. Int J Integ Biol. 2010
Zhang X, Azhar G, Chai J, Sheridan P, Nagano K, Brown T, Yang J, Khrapko K, Borras AM, Lawitts J, Misra RP, Wei JY: Cardiomyopathy in transgenic mice with cardiac-specific overexpression of serum response factor. Am J Physiol Heart Circ Physiol. 2001, 280 (4): H1782-1792.
Zhang X, Chai J, Azhar G, Sheridan P, Borras AM, Furr MC, Khrapko K, Lawitts J, Misra RP, Wei JY: Early postnatal cardiac changes and premature death in transgenic mice overexpressing a mutant form of serum response factor. J Biol Chem. 2001, 276 (43): 40033-40040. 10.1074/jbc.M104934200.
Schmittgen TD, Livak KJ: Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008, 3 (6): 1101-1108. 10.1038/nprot.2008.73.
Livak KJ, Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001, 25 (4): 402-408. 10.1006/meth.2001.1262.
Zhang X, Azhar G, Zhong Y, Wei JY: Identification of a novel serum response factor cofactor in cardiac gene regulation. J Biol Chem. 2004, 279 (53): 55626-55632. 10.1074/jbc.M405945200.
Zhang X, Azhar G, Huang C, Cui C, Zhong Y, Huck S, Wei JY: Alternative splicing and nonsense-mediated mRNA decay regulate gene expression of serum response factor. Gene. 2007, 400 (1-2): 131-139. 10.1016/j.gene.2007.06.008.
Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN: Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell. 2001, 105 (7): 851-862. 10.1016/S0092-8674(01)00404-4.
Zhang X, Azhar G, Zhong Y, Wei JY: Zipzap/p200 is a novel zinc finger protein contributing to cardiac gene regulation. Biochem Biophys Res Commun. 2006, 346 (3): 794-801. 10.1016/j.bbrc.2006.05.211.
Cullen BR: Transcription processing of human microRNA precursors. Mol Cell. 2004, 16 (6): 861-865. 10.1016/j.molcel.2004.12.002.
Shomron N, Levy C: MicroRNA-biogenesis and Pre-mRNA splicing crosstalk. J Biomed Biotechnol. 2009, 2009: 594678-
Fujita S, Ito T, Mizutani T, Minoguchi S, Yamamichi N, Sakurai K, Iba H: miR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J Mol Biol. 2008, 378 (3): 492-504. 10.1016/j.jmb.2008.03.015.
Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J: MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007, 116 (3): 258-267. 10.1161/CIRCULATIONAHA.107.687947.
Monzo M, Navarro A, Bandres E, Artells R, Moreno I, Gel B, Ibeas R, Moreno J, Martinez F, Diaz T, Martinez A, Balague O, Garcia-Foncillas J: Overlap** expression of microRNAs in human embryonic colon and colorectal cancer. Cell Res. 2008, 18 (8): 823-833. 10.1038/cr.2008.81.
D'Alessandra Y, Devanna P, Limana F, Straino S, Di Carlo A, Brambilla PG, Rubino M, Carena MC, Spazzafumo L, De Simone M, Micheli B, Biglioli P, Achilli F, Martelli F, Maggiolini S, Marenzi G, Pompilio G, Capogrossi MC: Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. 2010,
Latronico MV, Condorelli G: MicroRNAs and cardiac pathology. Nat Rev Cardiol. 2009, 6 (6): 419-429. 10.1038/nrcardio.2009.56.
van Rooij E, Olson EN: MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007, 117 (9): 2369-2376. 10.1172/JCI33099.
Krutzfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M: Silencing of microRNAs in vivo with 'antagomirs'. Nature. 2005, 438 (7068): 685-689. 10.1038/nature04303.
Gregory RI, Shiekhattar R: MicroRNA biogenesis and cancer. Cancer Res. 2005, 65 (9): 3509-3512. 10.1158/0008-5472.CAN-05-0298.
Denli AM, Tops BB, Plasterk RH, Ketting RF, Hannon GJ: Processing of primary microRNAs by the Microprocessor complex. Nature. 2004, 432 (7014): 231-235. 10.1038/nature03049.
Gabriely G, Wurdinger T, Kesari S, Esau CC, Burchard J, Linsley PS, Krichevsky AM: MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008, 28 (17): 5369-5380. 10.1128/MCB.00479-08.
Sayed D, Rane S, Lypowy J, He M, Chen IY, Vashistha H, Yan L, Malhotra A, Vatner D, Abdellatif M: MicroRNA-21 targets Sprouty2 and promotes cellular outgrowths. Mol Biol Cell. 2008, 19 (8): 3272-3282. 10.1091/mbc.E08-02-0159.
Selaru FM, Olaru AV, Kan T, David S, Cheng Y, Mori Y, Yang J, Paun B, ** Z, Agarwal R, Hamilton JP, Abraham J, Georgiades C, Alvarez H, Vivekanandan P, Yu W, Maitra A, Torbenson M, Thuluvath PJ, Gores GJ, LaRusso NF, Hruban R, Meltzer SJ: MicroRNA-21 is overexpressed in human cholangiocarcinoma and regulates programmed cell death 4 and tissue inhibitor of metalloproteinase 3. Hepatology. 2009, 49 (5): 1595-1601. 10.1002/hep.22838.
Cheng Y, Zhang C: MicroRNA-21 in cardiovascular disease. J Cardiovasc Transl Res. 2010, 3 (3): 251-255. 10.1007/s12265-010-9169-7.
Davis BN, Hilyard AC, Lagna G, Hata A: SMAD proteins control DROSHA-mediated microRNA maturation. Nature. 2008, 454 (7200): 56-61. 10.1038/nature07086.
Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ: The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006, 38 (2): 228-233. 10.1038/ng1725.
Bostjancic E, Zidar N, Stajer D, Glavac D: MicroRNAs miR-1, miR-133a, miR-133b and miR-208 Are Dysregulated in Human Myocardial Infarction. Cardiology. 2009, 115 (3): 163-169. 10.1159/000268088.
Matkovich SJ, Wang W, Tu Y, Eschenbacher WH, Dorn LE, Condorelli G, Diwan A, Nerbonne JM, Dorn GW: MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ Res. 2010, 106 (1): 166-175. 10.1161/CIRCRESAHA.109.202176.
Acknowledgements
This study was supported in part by NIH grants AG-018388, and AG-026091 from the Department of Health and Human Services, The Geriatric Research, Education, and Clinical Center (GRECC), and the Central Arkansas Veterans Healthcare System. We thank Dr. E. Olson for myocardin plasmid vector, and Dr. Y-H Zhou for her helpful input.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XMZ participated in the design of the study, carried out part of the experiments and drafted the manuscript. GA participated in the coordination of the study and the writing of the manuscript. SH participated in bioinformatics analysis. JYW led the study and participated in the design of the study, the statistical analysis and overall interpretation of results. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, X., Azhar, G., Helms, S.A. et al. Regulation of cardiac microRNAs by serum response factor. J Biomed Sci 18, 15 (2011). https://doi.org/10.1186/1423-0127-18-15
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1423-0127-18-15