
Abstract
SARS-CoV-2 is the virus responsible for the COVID-19 pandemic, and its effects on people worldwide continue to grow. Protein-targeted therapeutics are currently unavailable for this virus. As with other coronaviruses, the nucleocapsid (N) protein is the most conserved RNA-binding structural protein of SARS-CoV-2. The N protein is an appealing target because of its functional role in viral transcription and replication. Therefore, molecular docking method for structure-based drug design was used to investigate the binding energy and binding modes of various anti-N inhibitors in depth. The inhibitors selected were originally developed to target stress granules and other molecules involved in RNA biology, and were either FDA-approved or in the process of clinical trials for COVID-19. We aimed at targeting the N-terminal RNA binding domain (NTD) for molecular docking-based screening, on the basis of the first resolved crystal structure of SARS-CoV-2 N protein (PDB ID: 6M3M) and C-terminal domain (CTD) dimerization of the nucleocapsid phosphoprotein of SARS-COV-2 (PDB ID: 6WJI). Silmitasertib, nintedanib, ternatin, luteolin, and fedratinib were found to interact with RNA binding sites and to form a predicted protein interface with high binding energy. Similarly, silmitasertib, sirolimus-rapamycin, dovitinib, nintedanib, and fedratinib were found to interact with the SARS-CoV-2 N protein at its CTD dimerization sites, according to previous studies. In addition, we investigated an information gap regarding the relationships among the energetic landscape and stability and drug binding of the SARS-CoV-2 N NTD and CTD. Our in silico results clearly indicated that several tested drugs as potent putative inhibitors for COVID-19 therapeutics, thus indicating that they should be further validated as treatments to slow the spread of SARS-CoV-2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
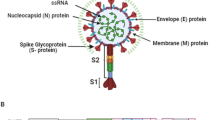
Since the emergence of the Delta variant of SARS-CoV-2 in May 2021, more than 155 million medically confirmed cases and approximately 3.24 million deaths occurred in more than 180 countries as a result of the COVID-19 pandemic [1]. The SARS-CoV-2/2019-nCoV novel coronavirus strain causes this respiratory disease [2, 3]. Repurposing of drugs has been a promising strategy at the forefront of strategies to address the continually growing number of COVID-19 cases [4,5,6,7,8]. In the current pandemic situation, drug repurposing should be considered a new avenue for the treatment of COVID-19 [5]. The coronavirus virion consists of structural proteins, namely spike (S), envelope (E), membrane (M), nucleocapsid (N) and, for some beta coronaviruses, hemagglutinin esterase [9]. The genomic structure of SARS-CoV-2 contains short untranslated regions and N-3′, E, M, 5′-replicase (rep)-S, identical to the genome structures of other coronaviruses [10]. As in other coronaviruses, the N protein is a crucial structural component of SARS-CoV-2 [11]. SARS-CoV-2 N has 90% sequence identity to the Severe Acute Respiratory Syndrome coronavirus N protein [12].
The N protein, which is naturally found within the virus, is the most conserved structural protein. It is required for viral replication and transcription, as it binds the viral RNA genome [13]. The N protein structure comprises an N-terminal RNA-binding domain (NTD), a C-terminal domain (CTD), and a naturally disordered central Arg/Ser-rich linker. For the SARS-CoV-2 N protein, each NTD molecule adopts a right-handed fist shape. The core sub domain consists of a five-stranded U-shaped antiparallel β-sheet with β4–β2–β3–β1–β5 topology, sandwiched between two short α-helices (α1 before the β2 strand and α2 after the β5 strand), and a protruding β-hairpin (β2′–β3′) is composed of mostly basic amino acid residues. New COVID-19 drug targets have been identified in the SARS-CoV-2-human interactome recently published by Gordon et al. [14]. Virtual screening and molecular docking have led to the identification of various inhibitors of SARS-CoV-2 [15].
Among the 332 interactions previously identified between viral and host proteins, most involve the innate immune signaling pathway [13, 16]. With this knowledge, researchers have identified an anti-N drug chain that shows excellent promise as a treatment for COVID-19 [17]. Through pathway analysis, a series of anti-N drugs with high potential to combat COVID-19 have been identified. Interestingly, some of the drugs target the N protein, which has been suggested to be a viable target for antiviral drug development [14, 16]. Yellow color indicates human drug target interaction with SARS-CoV-2 (Fig. 6A). G3BP1, G3BP2, and LARP1 human proteins interact with N of SARS CoV-2 and are drug response proteins. Host proteins are involved in RNA splicing, viral defense, ribosome biogenesis in eukaryotes, metabolism of RNA, and mRNA catabolic processes (Fig. 6B).

A Interactions between SARS-CoV-2 N and human proteins. B Bar graph of enriched terms across input gene lists, colored by p-values
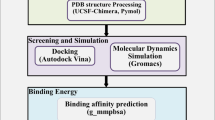
3.6 Identification of Potential Drug–Target Interactions
The STITCH database was used as a model for drug–protein interactions through a statistical approach. STITCH incorporates data from 2031 genomes on more than 5 million interactions between 430,000 chemicals and 9.6 million proteins. To predict protein–drug interactions, it primarily relies on keyword mining of the literature and experimental evidence. The likelihood that the expected relationship occurs is indicated by a confidence score (0–0.9). A confidence score of 0.9 or higher was used to discover the targets. To construct a network based on binding affinities (\({K}_{i}\) of protein–drug interactions with thickness of edges between nodes, increasing as \({K}_{i}\) value increases), we used STITCH, which is based on STRING v10 [38]. Figure 7 shows the STITCH predictions for the drug–gene relationships among the top five strongly binding drugs. Some of these drugs had relatively fewer high affinity binding targets. Prominent proteins included YES1, RET, FGR, and FGFR1/2/3, which are involved in cancer and associated pathways as well as endocytosis, and dovitinib has been found to target some of these proteins (Fig. 7A). Fedratinib interacted with the JAK2/JAK1 proteins, which are part of the JAK-STAT signaling pathway (Fig. 7B).

Drug–gene network constructed with STITCH. The edge width of protein–drug interactions is scaled according to the binding affinity between the drug and the protein. The drug–protein networks of A dovitinib, B fedratinib, C luteolin, D nintedanib, E rapamycin, and F silmitasertib
Luteolin, a flavone, had a low affinity for the proteins CASP3, JUN, CDK2, FOS, and MAPK8, which are involved in the TNF signaling pathway and cancer (Fig. 7C). However, certain drugs had several predicted protein interactions and interacted via various pathways. For example, nintedanib targets the Ras signaling pathway, cancer, and cytokine–cytokine receptor interaction proteins MAP3K7, JAK1, PDGFRA/B, LCK, KIT, MELK, FLT3, and KDR (Fig. 7D). Similarly, sirolimus/rapamycin, a kinase inhibitor, interacts with many targets in the mTOR and AMPK signaling pathways, and with proteins involved in immunosuppression, including mTOR, FKBP1A, and FKBP5 (Fig. 7E). Silmitasertib interacts with CSNK2A1, a serine/threonine kinase protein involved in cell cycle progression, apoptosis, transcription, and viral infection (Fig. 7A). Another drug, foretinib, is involved in endocytosis and focal adhesions, and has similar targets to nintedanib and dovitinib.
4 Discussion
As the RNA binding activity of N protein is essential for viral ribonucleoprotein formation and genome replication, blocking the RNA binding of the N-NTD has been demonstrated to be a potential treatment strategy. N protein is an essential RNA-binding protein with crucial roles in replication and transcription of viral RNA. An overall right-handed fold with a β-sheet core is found between loops, as revealed by recently solved crystal structures of the SARS-CoV-2 N-NTD (PDB ID: 6M3M) and C-CTD (PDB ID: 6WJI). The core region of the β-sheet consists of five antiparallel β-strands with a β6–β2–β5–β1–β7 topology flanked by a single short α-helix just before strand β2, and a protruding β-hairpin (β3 and β4) between strands β2 and β5. In addition, an NMR structure of the SARS-CoV-2 N-NTD in complex with RNA (PDB ID: 6YI3) suggested putative RNA binding sites of A51, T58, H60, R93, I95, L105, S106, R108, R150, and Y173. Moreover, the adenosine monophosphate (AMP) binding site has been structurally characterized in HCoV-OC43 N-NTD by Lin et al. [22]. N49, A51, S52, A56, R89, R108, Y110, Y112, and R150 compose the AMP binding site, as indicated by the structural superposition between the SARS-CoV-2 N-NTD and HCoV-OC43 N-NTD-AMP. Therefore, we extended our investigation by using structure-based molecular docking of N-NTD with different drugs to gain insights into the structural and molecular regions’ potential effectiveness in antiviral drug therapy. The investigated drugs included protein biogenesis inhibitors, anticancer compounds, antiinflammatory compounds, mTOR inhibitors, and stress granule modifiers. The docking results showed that 5 of the 20 drugs bound with strong binding affinity. Among N-NTD inhibitors, the key residues involved in binding were near the helix, i.e., Trp53, Ile75, Asn76, and Thr77; in the β3 strand, i.e., Arg93 and Arg94; and at the C-terminal interface, i.e., Ala153, Ala156, Ile158, Val159, and Gln161.
Silmitasertib is an antiviral drug that has been tested against the N protein of SARS-CoV-2, and found to block the CK2 and enhance SGs formation [39], thus inhibiting SARS-CoV-2 proliferation in vitro. Recently, Taiwan-headquartered Senhwa Biosciences Inc and the US National Institutes of Health (NIH) collaborated in analyzing the effectiveness of silmitasertib for the treatment of COVID-19 (https://www.biospectrumasia.com/news/34/15848/senhwa-biosciences-nih-to-co-develop-COVID-19-drug.html). The drug showed promise in controlling the proliferation of this RNA virus in human clinical tests. Silmitasertib was developed by Senhwa Biosciences to treat cancers, such as pediatric brain tumors, medulloblastoma, and bile duct cancer.
The second molecule tested was nintedanib, a tyrosine kinase inhibitor used to treat idiopathic pulmonary fibrosis or interstitial lung disease [40]. Very recently, the safety and efficacy of nintedanib ethanesulfonate have been analyzed in treating pulmonary fibrosis in patients with mild-to-extreme COVID-19. A placebo-controlled, a single-center, randomized study has been initiated and is currently in a phase 2 clinical trial (ClinicalTrials.gov identifier: NCT04338802).
In addition, the viral translation inhibitors ternatin and zotatifin, which is an FDA-approved drug for the treatment of multiple myeloma, have demonstrated the strongest binding affinity [41]. Plitidepsin is structurally similar to ternatin and is currently undergoing a clinical trial in COVID-19. The flavone luteolin, an antiinflammatory molecule, has broad antiviral properties [42, 43]. Previous studies have shown that luteolin inhibits SARS-CoV S protein and 3CL protease [44, 45]. Recently, both luteolin and quercetin have been identified through virtual screening and molecular docking as the best possible SARS-CoV-2 inhibitors [46, 47]. Furthermore, through SUMMIT, the world's most powerful supercomputer, high-throughput screening of small molecules interacting with the SARS-CoV-2 S protein or S protein–human ACE2 interface have recently been reported. Eriodictyol, a structural analog of luteolin, has been found to be a potential inhibitor of SARS-CoV-2 [48]. The last drug with considerable binding affinity toward N protein is fedratinib, an antiinflammatory JAK2 inhibitor. Wu et al. [49] have reported that fedratinib suppresses the expression of IL17, IL 22, and L23 in murine TH17 cells, and suggested that the drug may help mitigate the cytokine storm associated with SARS-CoV-2 infection. Stebbing et al. [50], through in silico artificial intelligence, have predicted significant beneficial effects of the antiinflammatory agents baricitinib, fedratinib, and ruxolitinib in the treatment of COVID-19.
The drug foretinib is an anticancer agent that inhibits vascular endothelial growth factor receptor (VEGFR) and hepatocyte growth factor receptor (HEGFR or MET) receptor. A recent study has reported that foretinib (DB12307) is a strong binder, on the basis of analysis through in silico virtual screening and molecular docking of 8548 ligands on the SARS-CoV-2 endoribonuclease NendoU (PDB ID: 6VWW) [51]. Our findings also have suggested that this drug binds the nucleocapsid protein with high binding affinity. Thus, foretinib may be repurposed as a broad-spectrum drug and tested against COVID-19 in the future.
Intriguingly, the docking of the N-NTD and C-CTD proteins revealed promising results for all five drugs tested. Notably, fedratinib and luteolin bind the ribonucleotide binding site and thus can inhibit RNA binding of the protein. Similarly, silmitasertib and nintedanib are positioned at the interface of two monomers and thus can impair the oligomerization of the protein. We recommend further experimental investigation of these compounds.
5 Conclusion
The highly immunogenic and abundant nature of the N protein makes it a novel target to treat infection of the respiratory system by SARS-CoV2. We extended the investigation of drug efficacy, stimulated by the recent SARS-CoV-2-host interactome and identification of several anti-N drugs, by using computational analysis. In this study, binding modes were chosen, and the most common anti-N drugs were selected. The probable molecular underpinnings of their effectiveness against COVID-19 have been identified. The docking results indicated that 5 of 20 anti-N inhibitors bind with the energetic landscape of a protein–drug complex and have high thermodynamic scores. The identified drugs have been shown to bind the ribonucleotide binding pocket and protein interface of the N-NTD and C-CTD, thereby suggesting mechanisms of action. Thus, the identification of compounds that bind the N-NTD and C-CTD and interfere with NTD–RNA and NTD–NTD interactions may assist in the development of broad-spectrum antiviral therapeutics.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Hu B, Guo H, Zhou P, Shi ZL. Characteristics of SARS-CoV-2 and COVID-19. Nat Rev Microbiol. 2021;19(3):141–54.
Mlcochova P, Kemp SA, Dhar MS, Papa G, Meng B, Ferreira IA, Datir R, Collier DA, Albecka A, Singh S, Pandey R. SARS-CoV-2 B. 1.617. 2 Delta variant replication and immune evasion. Nature. 2021;599(7883):114–9.
Sanayaima Singh RK, Zubbair Malik M, Brojen Singh RK. Diversity of SARS-CoV-2 isolates driven by pressure and health index. Epidemiol Infect. 2021;149: e38.
Rosa SGV, Santos WC. Clinical trials on drug repositioning for COVID-19 treatment. Rev Panam Salud Publica. 2020;44: e40.
Gupta RK, Nwachuku EL, Zusman BE, Jha RM, Puccio AM. Drug repurposing for COVID-19 based on an integrative meta-analysis of SARS-CoV-2 induced gene signature in human airway epithelium. PLoS One. 2021;16(9): e0257784.
Gupta V, Haider S, Verma M, Singhvi N, Ponnusamy K, Zubbair Malik M, Verma H, Kumar R, Sood U, Hira P, Satija S, Singh Y, Lal R. Comparative genomics and integrated network approach unveiled undirected phylogeny patterns, co-mutational hotspots, functional crosstalk and regulatory interactions in SARS-CoV-2. mSystems. 2021;6: e0003021.
Mishra CB, Pandey P, Sharma RD, Malik MZ, Mongre RK, Lynn AM, Prasad R, Jeon R, Prakash A. Identifying the natural polyphenol catechin as a multitargeted agent against SARSCoV-2 for the plausible therapy of COVID-19: an integrated computational approach. Brief Bioinform. 2021;22:1346–60.
Alam A, Khan A, Imam N, Siddiqui MF, Waseem M, Malik MZ, Ishrat R. Design of an epitope-based peptide vaccine against the SARS-CoV-2: a vaccine-informatics approach. Brief Bioinform. 2021;22:1309–23.
V’kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155–70.
Prajapat M, Sarma P, Shekhar N, Avti P, Sinha S, Kaur H, Kumar S, Bhattacharyya A, Kumar H, Bansal S, Medhi B. Drug targets for corona virus: a systematic review. Indian J Pharmacol. 2020;52(1):56–65.
Bai Z, Cao Y, Liu W, Li J. The SARS-CoV-2 nucleocapsid protein and its role in viral structure, biological functions, and a potential target for drug or vaccine mitigation. Viruses. 2021;13(6):1115.
Martínez YA, Guo X, Portales-Pérez DP, Rivera G, Castañeda-Delgado JE, García-Pérez CA, Enciso-Moreno JA, Lara-Ramírez EE. The analysis on the human protein domain targets and host-like interacting motifs for the MERS-CoV and SARS-CoV/CoV-2 infers the molecular mimicry of coronavirus. PLoS One. 2021;16(2): e0246901.
Chang CK, Lo SC, Wang YS, Hou MH. Recent insights into the development of therapeutics against coronavirus diseases by targeting N protein. Drug Discov Today. 2016;21(4):562–72.
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459–68.
Bhowmik D, Nandi R, Jagadeesan R, Kumar N, Prakash A, Kumar D. Identification of potential inhibitors against SARS-CoV-2 by targeting proteins responsible for envelope formation and virion assembly using docking based virtual screening, and pharmacokinetics approaches. Infect Genet Evol. 2020;84: 104451.
Banaganapalli B, Al-Rayes N, Awan ZA, Alsulaimany FA, Alamri AS, Elango R, Malik MZ, Shaik NA. Multilevel systems biology analysis of lung transcriptomics data identifies key miRNAs and potential miRNA target genes for SARS-CoV-2 infection. Comput Biol Med. 2021;135:104570.
Yang M, He S, Chen X, Huang Z, Zhou Z, Zhou Z, Chen Q, Chen S, Kang S. Structural insight into the SARS-CoV-2 nucleocapsid protein C-terminal domain reveals a novel recognition mechanism for viral transcriptional regulatory sequences. Front Chem. 2021;8:1238.
Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, **e X, ** Z, Peng J, Liu F, Li C. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–5.
Nakagawa K, Narayanan K, Wada M, Makino S. Inhibition of stress granule formation by Middle East respiratory syndrome coronavirus 4a accessory protein facilitates viral translation, leading to efficient virus replication. J Virol. 2018;92(20):e00902-e918.
Ivanov P, Kedersha N, Anderson P. Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol. 2019;11(5): a032813.
Chenavas S, Crepin T, Delmas B, Ruigrok RW, Slama-Schwok A. Influenza virus nucleoprotein: structure, RNA binding, oligomerization and antiviral drug target. Future Microbiol. 2013;8(12):1537–45.
Lin SY, Liu CL, Chang YM, Zhao J, Perlman S, Hou MH. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J Med Chem. 2014;57(6):2247–57.
Lo YS, Lin SY, Wang SM, Wang CT, Chiu YL, Huang TH, Hou MH. Oligomerization of the carboxyl terminal domain of the human coronavirus 229E nucleocapsid protein. FEBS Lett. 2013;587(2):120–7.
Monod A, Swale C, Tarus B, Tissot A, Delmas B, Ruigrok RW, Crepin T, Slama-Schwok A. Learning from structure-based drug design and new antivirals targeting the ribonucleoprotein complex for the treatment of influenza. Expert Opin Drug Discov. 2015;10(4):345–71.
Sarma P, Sekhar N, Prajapat M, Avti P, Kaur H, Kumar S, Singh S, Kumar H, Prakash A, Dhibar DP, Medhi B. In-silico homology assisted identification of inhibitor of RNA binding against 2019-nCoV N-protein (N terminal domain). J Biomol Struct Dyn. 2020;2020:1–11.
Malone B, Urakova N, Snijder EJ, Campbell EA. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat Rev Mol Cell Biol. 2022;23(1):21–39.
Kang S, et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm Sin B. 2020;10(7):1228–38.
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–61.
Dallakyan S, Olson AJ. Small-molecule library screening by docking with PyRx. Methods Mol Biol. 2015;1263:243–50.
Seeliger D, de Groot BL. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J Comput Aided Mol Des. 2010;24(5):417–22.
Laskowski RA, Swindells MB. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–86.
Szklarczyk D, Santos A, von Mering C, et al. STITCH 5: augmenting protein-chemical interaction networks with tissue and affinity data. Nucleic Acids Res. 2016;44:D380-384.
Tian W, Chen C, Lei X, et al. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:W363–7.
Jendele L, Krivak R, Skoda P, et al. PrankWeb: a web server for ligand binding site prediction and visualization. Nucleic Acids Res. 2019;47:W345–9.
Yang J, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics. 2013;29:2588–95.
Negi SS, Schein CH, Oezguen N, et al. InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics. 2007;23:3397–9.
Chen H, Zhou HX. Prediction of interface residues in protein-protein complexes by a consensus neural network method: test against NMR data. Proteins. 2005;61:21–35.
Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43:D447-452.
Reineke LC, Tsai WC, Jain A, Kaelber JT, Jung SY, Lloyd RE. Casein kinase 2 is linked to stress granule dynamics through phosphorylation of the stress granule nucleating protein G3BP1. Mol Cell Biol. 2017;37(4):e00596-e616.
Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, Richeldi L, Kolb M, Tetzlaff K, Stowasser S, Coeck C, Clerisme-Beaty E, Rosenstock B, Quaresma M, Haeufel T, Goeldner RG, Schlenker-Herceg R, Brown KK. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. 2019;381(18):1718–27.
Slaine PD, Kleer M, Smith NK, Khaperskyy DA, McCormick C. Stress granule-inducing eukaryotic translation initiation factor 4A inhibitors block influenza A virus replication. Viruses. 2017;9(12):388.
Fan W, Qian S, Qian P, Li X. Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res. 2016;220:112–6.
Yan H, Ma L, Wang H, Wu S, Huang H, Gu Z, Jiang J, Li Y. Luteolin decreases the yield of influenza A virus in vitro by interfering with the coat protein I complex expression. J Nat Med. 2019;73(3):487–96.
Yi L, et al. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J Virol. 2004;78(20):11334–9.
Jo S, Kim S, Shin DH, Kim MS. Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem. 2020;35(1):145–51.
Khaerunnisa S, Kurniawan H, Awaluddin R, Suhartati S. Potential inhibitor of COVID-19 main protease (Mpro) from several medicinal plant compounds by molecular docking study. Preprints. 2020;39(8):2000028.
Ton AT, Gentile F, Hsing M, Ban F, Cherkasov A. Rapid identification of potential inhibitors of SARS-CoV-2 main protease by deep docking of 1.3 billion compounds. Mol Inform. 2020;39(8):2000028.
Smith M. Repurposing therapeutics for COVID-19: supercomputer-based docking to the SARS-CoV-2 viral spike protein and viral spike protein-human ACE2 interface. Chem-Rxiv 2020.
Wu D, Yang XO. TH17 responses in cytokine storm of COVID-19: an emerging target of JAK2 inhibitor Fedratinib. J Microbiol Immunol Infect. 2020;53(3):368–70.
Stebbing J, Krishnan V, de Bono S, et al. Mechanism of baricitinib supports artificial intelligence-predicted testing in COVID-19 patients. EMBO Mol Med. 2020;12:e12697. https://doi.org/10.15252/emmm.202012697.
Kim Y, Jedrzejczak R, Maltseva N, Endres M, Godzik A, Michalska K, Joachimiak A. Crystal structure of NSP15 endoribonuclease from SARS CoV-2. Protein Sci. 2020;29(7):1596–605.
Funding
M.S. was financially supported by the Dr Sulaiman Al Habib Research Center under an Excellence Award (Project Reference Number: COVID-19 RC#09).
Author information
Authors and Affiliations
Contributions
ARS, MS, and SI: study conceptualization and writing (review and editing) the manuscript. RA and HA: data curation, formal analysis, and writing (original draft). MS: funding acquisition and project administration. SI and MS: supervision of the project. ARS, RA, HA, LV, and MS: formal analysis and writing (original draft).
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare they have no conflicts of interest.
Ethical approval
No ethical issues exist in the article, and all authors approve publication.
Consent for publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Afreen, R., Iqbal, S., Shah, A.R. et al. In Silico Identification of Potential Inhibitors of the SARS-CoV-2 Nucleocapsid Through Molecular Docking-Based Drug Repurposing. Dr. Sulaiman Al Habib Med J 4, 64–76 (2022). https://doi.org/10.1007/s44229-022-00004-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44229-022-00004-z