
Abstract
Cardio-renal syndromes (CRS) are defined as disorders of the heart and kidney whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other. CRS have been classified into five categories, where types 2 and 4 represent respectively chronic cardio-renal and chronic reno-cardiac syndromes. In these conditions, the chronic disorder of either the heart or kidney has been shown to induce some degree of cachexia. At the same time, cachexia has been proposed as a possible mechanism contributing to the worsening of such pathological organ cross talk. Common pathogenetic mechanisms underlie body wasting in cachectic states of different chronic heart and kidney diseases. In these circumstances, a vicious circle could arise, in which cachexia associated with either heart failure or chronic kidney disease may contribute to further damage of the other organ. In chronic CRS, activation of the immune and neuroendocrine systems contributes to the genesis of cachexia, which in turn can negatively affect the heart and kidney function. In patients with cardiac sustained activation of the immune and neuroendocrine systems and oxidative stress, renal vascular resistance can increase and therefore impair renal perfusion, leading to worsening kidney function. Similarly, in renal cachexia, increased levels of pro-inflammatory cytokines can cause progressive left ventricular systolic dysfunction, myocardial cell death, endothelial dysfunction and increased myocardial fibrosis, with consequent impairment of the chronic reno-cardiac syndrome type 4. Thus, we speculate that the occurrence of different types of chronic CRS could represent a fundamental step in the genesis of cachexia, being renal and cardiac dysfunction closely related to the occurrence of systemic disorders leading to a final common pathway. Therefore, the heart and kidney and cachexia represent a triad causing a vicious circle that increases mortality and morbidity: In such circumstances, we may plausibly talk about cardio-renal cachexia syndrome. Complex interrelations may explain the transition from CRS to cachexia and from cachexia to CRS. Identification of the exact mechanisms occurring in these conditions could potentially help in preventing and treating this deadly combination.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Heart failure (HF) is a clinical condition associated with poor prognosis and high hospitalisation rates due to exacerbation of the disease [1, 2]. Concomitant kidney abnormalities are frequent: Only 7% of chronic HF patients have a preserved kidney function, with the majority of patients having mild to moderate impairment of glomerular filtration rate [3]. Furthermore, acute HF syndromes are frequently complicated by acute kidney injury (AKI) [4]. In both chronic and acute settings of HF, many studies have shown that worsening renal function is associated with dismal prognosis [4–6]. These conditions are generally grouped under the umbrella term cardio-renal syndromes (CRS).
Cachexia is a frequent and serious complication of chronic diseases, including HF, chronic kidney disease (CKD), cancer and acquired immunodeficiency syndrome [7, 8]. It has been calculated that in 2006, approximately 5 million people in the USA were cachectic because of cancer or other chronic diseases [9]. Uremic cachexia increases the risk of death of patients with CKD up to 100- to 200-fold that of the general population [10]; similarly, cardiac cachexia is a negative prognostic factor in patients with CHF independently of left ventricular systolic function and exercise capacity: cachectic patients have a poor outcome [11, 12], with survival rates at 18 months <50%.
Interestingly, cardio-renal syndromes and cachexia share common pathophysiological mechanisms, and we hypothesise that the presence of one of these conditions may favour the occurrence or worsening of the other. A better understanding of the complex interplay between the heart and kidney could help improve the management of these patients. The same may be true for cachexia. In the present article, we will focus on the relationship between cachexia and cardio-renal syndromes, exploring the potential mechanisms contributing to the vicious circle leading to a combined cardio-renal cachexia syndrome (CRCS) and its progressive worsening.
2 Cardio-renal syndromes
CRS, defined as ‘disorders of the heart and kidney whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other’ [13], have been recently classified into five categories [14].
CRS type 1, or acute CRS, is determined by any of the acute HF syndromes [2] causing AKI. This type of CRS is frequent, with acute HF syndromes being the most common and the most expensive diagnosis-related group for Medicare patients [15] and worsening renal function occurring in 30–45% of patients during hospitalisation [16, 17]. The mechanisms contributing to worsening renal function after an episode of acute HF are multiple and complex. Whilst some haemodynamic factors, like heart rate, systolic arterial blood pressure and pulmonary wedge pressure, do not seem to play a major role, an incremental risk of develo** AKI has been described with increased central venous pressure [18]. Other factors may contribute to worsening renal function in acute HF, like drug and contrast media use, or systemic activation of neurohormonal systems and pro-inflammatory cytokines [19–21].
CRS type 2, or chronic CRS, is defined as progressive CKD caused by chronic cardiac dysfunction. The prevalence of this subtype is approximately 25% [22], and the associated risk of cardiovascular mortality and hospitalisation for worsening HF for chronic HF patients is more than three times higher when the glomerular filtration rate (GFR) falls below 45 ml min−1/1.73 m2 as compared with a normal GFR. The pathophysiological basis for the interactions between heart and kidney in CRS type 2 has not been fully understood yet. It is likely that a chronic hypoperfusion of the kidney may progressively lead to alterations in renal vasculature; increased renal vascular resistance, continuous activation of hormonal systems and drug-induced damage all contribute to further impairment of renal function moving from an initial insult to a progressive fibrosis and sclerosis of the renal parenchyma.
CRS type 3, or acute reno-cardiac syndrome, is characterised by an AKI which leads to acute HF, ischaemia or arrhythmias. AKI can affect the heart through multiple mechanisms. First, volume overload can lead to pulmonary congestion, particularly in the presence of left ventricular dysfunction; second, acidosis and uraemia itself can depress cardiac systolic function [23, 24], and acidosis and hyperkalemia carry a pro-arrhythmic risk. Another important factor is the activation of inflammation at the cardiac level, which can be triggered by renal ischaemia [19, 25, 26].
CRS type 4, or chronic reno-cardiac syndrome, is defined as a condition in which primary CKD contributes to cardiac abnormalities of various types and degrees, like hypertrophy, diastolic and systolic dysfunction, and increased risk of cardiovascular morbidity and mortality [27, 28]. Many pathways are implicated as pathogenetic mechanisms of cardiac damage in chronic renal disease: Activation of the renin–angiotensin–aldosterone system, volume overload, endothelial dysfunction, inflammation and anaemia all can contribute to functional and structural changes in the heart and vessels [29]. Less than 20% of patients with end-stage renal disease have a normal heart at echocardiography, whilst the majority have left ventricular hypertrophy or dilation [30]. A baseline GFR <60 ml min−1/1.73 m2 is independently associated with increased incidence of peripheral artery disease [31] and CHF [32], and almost half of the mortality affecting patients with CKD is due to cardiovascular events [33, 34].
CRS type 5, or secondary CRS, occurs when an acute or chronic systemic disorder causes both renal and cardiac dysfunction. Examples of this type of CRS include sepsis, haemorrhagic shock, diabetes, sarcoidosis and amyloidosis. The model of sepsis is paradigmatic in this context: Up to 60% of patients with systemic infection develop AKI, which contributes to higher morbidity and mortality [35, 36]; at the same time, cardiac damage is common. Septic patients have increased markers of cardiac-specific damage [37], and activation of the inflammatory response is directly responsible for cardiac cell apoptosis [38] and depressed left ventricular systolic function [39]. These abnormalities can trigger a vicious circle in which impairment of one organ can negatively affect the function of the other.
3 Cachexia in chronic diseases: definition and epidemiology
Cachexia is a complex syndrome associated with poor outcome, independently of the underlying primary disease. It is characterised by loss of skeletal muscle with or without loss of fat mass. Several definitions have been proposed, most of them focusing on the dominant feature of weight loss alone, and only a minority of authors recognised the importance of body composition changes over time. Other causes of weight loss include anorexia, sarcopenia and malnutrition, all of them possibly occurring both in HF and in CKD. Differentiation from other syndromes of weight loss is important to the early recognition and possible management of the syndrome. Carr et al. [40] defined cachexia as a body fat content of <15% in men and <22% in women, or a percentage of ideal weight <90%, whilst later cachexia was defined as a loss of >10% of lean tissue [41]. Nevertheless, these definitions depend on an evaluation of body composition, which is frequently not applicable in clinical practice. A previous definition, derived from the SOLVD database, appears to be the most appropriate to use in clinical practice since it does not require biochemical or skeletal muscle performance tests. Cachexia should therefore be defined as ‘a non-oedematous weight loss of >6% of total body weight over a period of 6 or more months’ [12].
More recently, a new definition has been proposed, which takes into account the concomitant clinical features associated with weight loss; according to the definition proposed by Evans et al. [42], cachexia is ‘a weight loss of 5% in twelve months or less (or a body mass index <20 kg/m2) in a patient with underlying chronic disease and at least three of the following criteria: decreased muscle strength, fatigue, anorexia, low fat-free mass index and abnormal biochemistry (inflammation, anaemia, low serum albumin)’ (Table 1).
Cachexia is frequently observed in association with many chronic disorders. Up to 60% of patients with end-stage renal disease (ESRD) present with malnutrition [43, 44], where this term does not only imply reduced energy intake, like in simple starvation, but also complex metabolic changes. This entity has been identified as malnutrition–inflammation–atherosclerosis (MIA) syndrome with potential genetic implications in its pathogenesis [45]. The prevalence of cardiac cachexia, in one of our initial studies, was 16% [11]; subsequent observations [12] reported an incidence of cardiac cachexia of 35%. Interestingly, in recent years, an inverse association with all-cause mortality and BMI in patients on haemodialysis has been reported [46], and preliminary studies suggest that this ‘reverse epidemiology’ is also present in CKD patients who are not on dialysis [47], particularly in proteinuric CKD [48]. Paradoxically, patients with higher BMI have lower all-cause and cardiovascular mortality. The occurrence of cachexia has therefore important prognostic implications in CKD, similarly to other chronic diseases; the understanding of the mechanisms that lead to cachexia might therefore help in improving the outcome of CKD patients.
4 Pathophysiological mechanisms of cachexia syndrome
4.1 Chronic HF
In 1984, Braunwald [49] suggested that in cardiac cachexia, right ventricular failure could be predominant; interestingly, the only predictor of malnutrition in patients with severe CHF was an increased right atrial pressure [40]. In another study, cardiac output and systolic and diastolic arterial pressures were found to be significantly lower in cachectic patients as compared with non-cachectic patients [50]. These findings suggest that haemodynamic conditions may play a role in the genesis of cardiac cachexia. Nevertheless, the most important players are neurohormonal and immune mechanisms. The trigger for immune activation has been reported to be pressure overload [51] and splanchnic venous congestion with consequent bacterial translocation from the gut into the bloodstream [52]. Levine et al. [53] were the first to demonstrate increased levels of tumour necrosis factor-alpha in cachectic patients. Other studies confirmed a marked immune activation in patients with cardiac cachexia [54] and the expression of cytokines in the myocardium of CHF patients [55]. Pro-inflammatory cytokines are the major determinants of catabolism, and we have previously shown that circulating tumour necrosis factor-alpha, originally called ‘cachectin’, is the strongest predictor of the degree of previous weight loss in cachectic patients [56] and correlates with a poor prognosis [57]. Immune activation in HF patients can cause worsening cardiac function [58] through the induction of apoptosis [59] and nitric oxide downregulation [60], impaired exercise capacity [61] and skeletal muscle abnormalities [61, 62]. Cytokines also reduce constitutive nitric oxide synthase mRNA in endothelial cells [63] and therefore impair endothelial function. In fact, an inverse relationship between peripheral blood flow and TNF levels has been reported in chronic HF patients [64].
The third key mechanism in the development of cardiac cachexia is neurohormonal activation. Increased tone of the adrenergic and renin–angiotensin–aldosterone systems aim to maintain cardiac output, but at the expense of increased vascular resistance and afterload, with further left ventricular remodelling and dysfunction. Interestingly, the same hormonal systems play an important role in peripheral tissue wasting: Angiotensin II and catecholamines have pro-apoptotic effects and increase resting metabolic rate [65, 66]. Similarly, abnormalities in the growth hormone axis and cortisol systems have been reported in chronic HF patients [67, 68]. Taken together, these abnormalities lead to an imbalance between the catabolic and anabolic states, confirmed by the observation of significantly higher levels of catabolic hormones in cachectic as compared with non-cachectic patients [53], with consequent muscle wasting, a dominant feature of cachectic patients [69]. In fact, muscle weakness and early fatigue are two of the main symptoms of HF patients [70, 71] and occur most commonly in patients with advanced HF or cachexia [72].
Finally, regulation of feeding and energy balance may be altered in chronic HF patients, in particular in patients with cardiac cachexia. Ghrelin, mainly produced by the stomach fundus, induces the release of growth hormone and regulates appetite [73, 74]. It has been reported to be increased in cachectic patients [75], and the increase in ghrelin was correlated with the increase in circulating growth hormone; it may therefore have a role in attenuating the catabolic–anabolic imbalance via growth hormone-releasing effects. Insulin and growth hormone resistance may occur secondary to HF [76, 77], with consequent loss of their anabolic effects.
4.2 Chronic kidney disease
CKD and ESRD are frequently complicated by progressive deterioration of nutritional status [8], which significantly affects prognosis and quality of life. Cachexia in CKD, similarly to HF, is a complex syndrome which derives not only by a reduced energy intake but is also the consequence of multiple pathogenetic mechanisms, including increased protein catabolism, reduced anabolism, increased energy expenditure and loss of adipose tissue. A variety of terms have been used to define this syndrome in CKD, like protein-energy wasting [78], malnutrition–inflammation complex syndrome [79] and kidney disease wasting [80], underlying the role of different pathways. As mentioned above, the accelerated cardio-renal syndrome type 4 in malnourished patients has also been defined as MIA syndrome based on the relation among malnutrition, inflammation and accelerated atherosclerosis. A major determinant of the disease process in CKD is anorexia. Amongst patients on renal replacement therapy, impaired appetite is reported in 38% and is associated with worse morbidity and mortality [81, 82]. Interestingly, anorexia was associated with increased inflammatory markers, suggesting a possible role for immune activation in regulating the stimuli for food intake. Furthermore, similarly to HF, it has been repeatedly reported that inflammation is closely related to qualitative and quantitative changes in the skeletal muscles of patients with CKD [83, 84]. Activation of the protease caspase-3 is the initial step of muscle protein degradation, and this inflammation-mediated proteolytic pathway is not counterbalanced by increased anabolic pathways in CKD [83]. Growth hormone (GH) resistance, due to the reduced density of GH receptors and abnormal intracellular signalling, contributes to increased skeletal muscle protein catabolism [80] and failure of anabolism in patients with end-stage renal disease.
Increased protein breakdown in CKD is also associated with insulin resistance [85], which frequently occurs in end-stage renal disease [86] and is triggered by inflammation through an increase in oxidative stress [87]. Therefore, inflammation can contribute to the disease process directly, activating proteolysis, and indirectly, through insulin resistance and oxidative stress. IN CKD, an increase in C-reactive protein is common as it is a decrease of acute phase proteins such as albumin. The mechanism that leads to inflammation in ESRD is complex and not fully understood yet, but recent studies paid attention on the gut as a possible source of cytokine stimulus [88]. In states of intestinal under perfusion, as during haemodialysis [89], the permeability of the intestinal wall is increased as a result of hypoxia and local production of lipopolysaccharide (LPS) [90], allowing LPS leakage into the bloodstream, with consequent immune activation [91]. These complex interactions between gut and kidney, for which the term ‘intestinal–renal syndrome’ has been proposed [91], could explain the occurrence of cardiovascular abnormalities during dialysis, like arrhythmias, silent ischaemia [92] and myocardial stunning [93], through the production of pro-inflammatory cytokines with consequent cardiac and vascular dysfunction [60, 94]. Further mechanisms have been indicated as potential causes of accelerated muscle wasting and intestinal abnormalities such as the lack of vitamin D receptor activation [95] and the lack of the anti-apoptotic effect at tissue levels of erythropoietin [96]; both conditions are typical of CKD and tend to worsen with progression of renal dysfunction.
Finally, abnormalities in ghrelin system have been reported in patients with CKD. Plasma ghrelin levels increase in dialysis patients [97] because of reduced ghrelin degradation—which occurs mainly in the kidney [98]. Two major forms of circulating ghrelin are acylated and des-acyl ghrelin. Acylated ghrelin stimulates food intake [99], downregulates pro-inflammatory cytokines [100] and improves lean body mass [101], whilst the des-acyl form induces a negative energy balance and could be involved in anorexia in CKD patients [98]. The different effects of the two forms can explain the conflicting results of previous studies on the exact role of this peptide on energy metabolism and regulation of food intake.
5 The cardio-renal cachexia syndrome: a cachexia-mediated negative heart–kidney cross talk
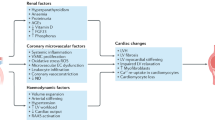
Chronic HF and CKD can be both complicated by the occurrence of cachexia, and interestingly, the underlying pathophysiological mechanisms are similar in the two conditions, as reviewed in the present article. Thus, since chronic heart and kidney interactions are at the base of CRS types 2 and 4, we could propose a new entity called cardio-renal cachexia syndrome (CRCS). It had been previously suggested that common pathogenetic mechanisms underlie body wasting in cachectic states of different chronic heart and kidney diseases [102]. Now we hypothesise that a vicious circle could arise, in which cachexia associated with either HF or CKD may contribute to further damage of the other organ (Fig. 1). Activation of the immune and neuroendocrine systems contribute to the genesis of cachexia, which in turn can negatively affect the heart and kidney function, worsening CRS types 2 and 4. In patients with cardiac cachexia, the same endogenous factors can affect different target organs other than the heart, particularly the kidney. Sustained activation of the immune and neuroendocrine systems and oxidative stress can increase renal vascular resistance and therefore impair renal perfusion, leading to worsening kidney function, i.e. a CRS type 2, or chronic cardio-renal syndrome. Similarly, in renal cachexia, increased levels of pro-inflammatory cytokines can cause progressive left ventricular systolic dysfunction, myocardial cell death, endothelial dysfunction and increased myocardial fibrosis [103], with consequent impairment of the haemodynamic conditions corresponding to the chronic reno-cardiac syndrome, or CRS type 4.

Cardio-renal cachexia syndromes
Furthermore, we speculate that the occurrence of different types of CRS could also represent a fundamental step in the genesis of cachexia, being renal and cardiac dysfunction closely related to the occurrence of systemic congestion, a central player in the theory of LPS translocation and immune activation, and to neurohormonal activation, with consequent catabolic/anabolic imbalance, as previously discussed.
In conclusion, the heart and kidney are players of the same game at different levels, and when cachexia is involved, we may plausibly talk about CRCS. Complex interrelations may explain the transition from CRS to cachexia and from cachexia to CRS. Identification of the exact mechanisms occurring in these conditions could potentially help in preventing and treating this deadly combination.
References
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics—2010 update: a report from the American Heart Association. Circulation. 2010;121:e46–e215.
Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol. 2009;53:557–73.
de Silva R, Nikitin NP, Witte KK, Rigby AS, Goode K, Bhandari S, et al. Incidence of renal dysfunction over 6 months in patients with chronic heart failure due to left ventricular systolic dysfunction: contributing factors and relationship to prognosis. Eur Heart J. 2006;27:569–81.
Heywood JT, Fonarow GC, Costanzo MR, Mathur VS, Wigneswaran JR, Wynne JR. High prevalence of renal dysfunction and its impact on outcome in 118,465 patients hospitalized with acute decompensated heart failure: a report from the ADHERE database. J Card Fail. 2007;13:422–30.
Bibbins-Domingo K, Lin F, Vittinghoff E, Barrett-Connor E, Grady D, Shlipak MG. Renal insufficiency as an independent predictor of mortality among women with heart failure. J Am Coll Cardiol. 2004;44:1593–600.
Hillege HL, Nitsch D, Pfeffer MA, Swedberg K, McMurray JJ, Yusuf S, et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–8.
Tisdale MJ. Biology of cachexia. J Natl Cancer Inst. 1997;89:1763–73.
Morley JE, Thomas DR, Wilson MM. Cachexia: pathophysiology and clinical relevance. Am J Clin Nutr. 2006;83:735–43.
Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev. 2009;89:381–410.
Mak RH, Cheung W. Energy homeostasis and cachexia in chronic kidney disease. Pediatr Nephrol. 2006;21:1807–14.
Anker SD, Ponikowski P, Varney S, Chua TP, Clark AL, Webb-Peploe KM, et al. Wasting as independent risk factor for mortality in chronic heart failure. Lancet. 1997;349:1050–3.
Anker SD, Negassa A, Coats AJ, Afzal R, Poole-Wilson PA, Cohn JN, et al. Prognostic importance of weight loss in chronic heart failure and the effect of treatment with angiotensin-converting enzyme inhibitors: an observational study. Lancet. 2003;361:1077–83.
Ronco C, House AA, Haapio M. Cardiorenal syndrome: refining the definition of a complex symbiosis gone wrong. Intensive Care Med. 2008;34:957–62.
Ronco C, Haapio M, House AA, Anavekar N, Bellomo R. Cardiorenal syndrome. J Am Coll Cardiol. 2008;52:1527–39.
Rosamond W, Flegal K, Furie K, et al. Heart disease and stroke statistics—2008 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2008;117:e25–e146.
Nohria A, Hasselbad V, Stebbins A, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. 2008;51:1268–74.
Klein L, Massie BM, Leimberger JD, et al. Admission or changes in renal function during hospitalization for worsening heart failure predict postdischarge survival: results from the Outcomes of a Prospective Trial of Intravenous Milrinone for Exhacerbation of Chronic Heart Failure (OPTIME-CHF). Circulation: Heart Fail. 2008;1:25–33.
Mullens W, Abrahams Z, Francis GS, Soko G, Taylo DO, Starling RG, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. 2009;53:589–696.
Berl T, Henrich W. Kidney–heart interactions: epidemiology, pathogenesis, and treatment. Clin J Am Soc Nephrol. 2006;1:8–18.
Hasselblad V, Gattis Stough W, Shah MR, et al. Relation between dose of loop diuretics and outcomes in a heart failure population: results of the ESCAPE trial. Eur J Heart Fail. 2007;9:1064–9.
McCullough PA. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419–28.
Hillege HL, Nitsch D, Pfeffer MA, et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–8.
Blake P, Hasegawa Y, Khosla MC, Fouad-Tarazi F, Satura N, Paganini EP. Isolation of ‘myocardial depressant factor(s)’ from the ultrafiltrate of heart failure patients with acute renal failure. ASAIO J. 1996;42:M911–5.
Brady JP, Hasbargen JA. A review of the effects of correction of acidosis on nutrition in dialysis patients. Semin Dial. 2000;13:252–5.
Kelly KJ. Acute renal failure: much more than a kidney disease. Semin Nephrol. 2006;26:105–13.
Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003;14:1549–58.
Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112–9.
Go AS, Chertow GM, Fan D, McCulloch E, Hsu CY. Chronic kidney disease and the risk of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–305.
McCollough PA. Why is chronic kidney disease the ‘spoiler’ for cardiovascular outcomes? J Am Coll Cardiol. 2003;41:725–8.
Parfrey PS, Foley RN, Harnett JD, Kent CM, Muraay DC, Barre PE. Outcome and risk factors for left ventricular disorders in chronic uremia. Nephrol Dial Transplant. 1996;11:1277–85.
Wattanakit K, Folsom AR, Selvin E, Coresh J, Hirsch AT, Weatherley BD. Kidney function and risk of peripheral arterial disease: results from the Atherosclerosis Risk in Communities (ARIC) Study. J Am Soc Nephrol. 2007;18:629–36.
Kottgen A, Russell SD, Loehr LR, et al. Reduced kidney function as a risk factor for incident heart failure: the Atherosclerosis Risk in Communities (ARIC) Study. J Am Soc Nephrol. 2007;18:1307–15.
Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–63.
Tonelli M, Wiebe N, Culleton B, et al. Chronic kidney disease and mortality risk: a systematic review. J Am Soc Nephrol. 2006;17:2034–47.
Hoste EA, Lameire NH, Vanholder RC, Benoit DD, Decruyenaere JM, Colardyn FA. Acute renal failure in patients with sepsis in a surgical ICU: predictive factors, incidence, comorbidity and outcome. J Am Soc Nephrol. 2003;14:1022–30.
Vincent JL, Sakr Y, Sprung CL, Ranieri VM, Reinhart K, Gerlach H, et al. Sepsis in European intensive care units: results of the SOAP Study. Crit Care Med. 2006;34:344–53.
Turner A, Tsamitros M, Bellomo R. Myocardial cell injury in septic shock. Crit Care Med. 1999;27:1775–80.
Pulkki KJ. Cytokines and cardiomyocyte death. Ann Med. 1997;29:339–43.
Bozkurt B, Kribbs SB, Clubb Jr FJ, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumour necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;98:149–56.
Carr JG, Stevenson LW, Walden JA, Heber D. Prevalence and haemodynamic correlates of malnutrition in severe congestive heart failure secondary to ischaemic or idiopathic dilated cardiomyopathy. Am J Cardiol. 1989;63:709–13.
Freeman LM, Roubenoff R. The nutritional implications of cardiac cachexia. Nutr Review. 1994;52:340–7.
Evans WJ, Morley JE, Argilès J, Bales C, Baracos V, Guttridge D, et al. Cachexia: a new definition. Clin Nutr. 2008;27:793–9.
Kalanthar-Zadeh K, Balakrishnan VS. The kidney disease wasting: inflammation, oxidative stress, and diet–gene interaction. Hemodyalisis Int. 2006;10:315–25.
Kalanthar-Zadeh K, Ikizler TA, Block G, Avram MM, Kopple JD. Malnutrition–inflammation complex syndrome in dialysis patients: causes and consequences. Am J Kidney Dis. 2003;42:864–81.
Pecoits-Filho R, Nordfors L, Lindholm B, Hoff CM, Schalling M, Stenvinkel P. Genetic approaches in the clinical investigation of complex disorders: malnutrition, inflammation, and atherosclerosis (MIA) as a prototype. Kidney Int Suppl. 2003;84:S162–7.
Herselman M, Esau N, Kruger JM, Labadarios D, Moosa MR. Relationship between body mass index and mortality in adults on maintenance hemodialysis: a systematic review. J Ren Nutr. 2010;20:281–92.
Kovesdy CP, Anderson JE. Reverse epidemiology in patients with chronic kidney disease who are not yet on dialysis. Semin Dial. 2007;20:566–9.
Obermayr RP, Temml C, Gutjahr G, Kainz A, Klauser-Braun R, Fugger R, et al. Body mass index modifies the risk of cardiovascular death in proteinuric chronic kidney disease. Nephrol Dial Transplant. 2009;24:2421–8.
Braunwald E. Clinical manifestation of heart failure. In: Braunwald E, editor. Heart disease. A textbook of cardiovascular medicine, vol. 1. Philadelphia: Saunders; 1984. p. 499.
MacGowan G, Mann D, Kormos R, Feldman A, Murali S. Circulating interleukin-6 in severe heart failure. Am J Cardiol. 1997;79:1128–31.
Kapadia SR, Oral H, Lee J, Nakano M, Taffet GE, Mann DL. Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ Res. 1997;81:187–95.
Niebauer J, Volk HD, Kemp M, et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet. 1999;353:1838–42.
Levine B, Kalman J, Mayer L, Fillit H, Packer M. Elevated circulating levels of tumour necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323:236–41.
Dutka DP, Elborn JS, Delamere F, Shale DJ, Morris GK. Tumour necrosis factor alpha in severe congestive cardiac failure. Br Heart J. 1993;70:141–3.
Torre-Amione G, Kapadia S, Lee J, et al. Tumor necrosis factor-alpha and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–11.
Anker SD, Chua TP, Ponikowski P, et al. Hormonal changes and catabolic/anabolic imbalance in chronic heart failure and their importance for cardiac cachexia. Circulation. 1997;96:526–34.
Rauchhaus M, Doehner W, Francio DP, et al. Plasma cytokine parameters and mortality in patients with chronic heart failure. Circulation. 2000;102:3060–7.
Bozkurt B, Kribbs S, Clubb Jr FJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-α promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97:1382–91.
Krown KA, Page MT, Nguyen C, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes; involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. 1996;98:2854–65.
Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. 1992;257:387–9.
Cicoira M, Bolger AP, Doehner W, Rauchhaus M, Davos C, Sharma R, et al. High tumour necrosis factor-alpha levels are associated with exercise intolerance and neurohormonal activation in chronic heart failure patients. Cytokine. 2001;15:80–6.
Anker SD, Ponikowski P, Clark AL, Leyva F, Rauchhaus M, Kemp M, et al. Cytokines and neurohormones relating to body composition alterations in the wasting syndrome of chronic heart failure. Eur Heart J. 1999;20:683–93.
Yoshizumi M, Perrella MA, Burnett JC, Lee ME. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circ Res. 1993;73:205–9.
Anker SD, Volterrani M, Egerer KR, et al. Tumour necrosis factor alpha as a predictor of impaired peak leg blood flow in patients with chronic heart failure. Q J Med. 1998;91:199–203.
Kajstura J, Cigola E, Malhotra A, et al. Angiotensin II induces apoptosis of adult ventricular myocytes in vitro. J Mol Cell Cardiol. 1997;29:859–70.
Obisesan TO, Toth MJ, Yki-Jarvinen H. Free-fatty acid kinetics and oxidation in congestive heart failure. Am J Cardiol. 1996;77:1250–2.
Anand IS, Ferrari R, Kalra GS, Wahi PL, Poole-Wilson PA, Harris PC. Edema of cardiac origin. Studies of body water and sodium, renal function, hemodynamic indexes, and plasma hormones in untreated congestive cardiac failure. Circulation. 1989;80:299–305.
Colao A. The GH–IGF-I axis and the cardiovascular system: clinical implication. Clin Endocrinol. 2008;69:347–58.
Anker SD, Swan JW, Volterrani M, et al. The influence of muscle mass, strength, fatigability and blood flow on exercise capacity in cachectic and non-cachectic patients with chronic heart failure. Eur Heart J. 1997;18:259–69.
Coats AJ, Clark AL, Piepoli M, Volterrani M, Poole-Wilson PA. Symptoms and quality of life in heart failure: the muscle hypothesis. Br Heart J. 1994;72:S36–9.
Cicoira M, Zanolla L, Franceschini L, Rossi A, Golia G, Zamboni M, et al. Skeletal muscle mass independently predicts peak oxygen consumption and ventilatory response to exercise in chronic heart failure patients. J Am Coll Cardiol. 2001;37:1808–12.
Harrington D, Anker SD, Chua TP, Webb-Peploe KM, Ponikowski P, Poole-Wilson PA, et al. Skeletal muscle function and its relation to exercise tolerance in chronic heart failure. J Am Coll Cardiol. 1997;30:1758–64.
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–60.
Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, et al. Ghrelin causes hyperphagia and obesity in rats. Diabetes. 2001;50:2540–7.
Nagaya N, Uematsu M, Kosjima M, Date Y, Nakazato M, Okumura H, et al. Elevated circulating level of ghrelin in cachexia associated with chronic heart failure. Relationships between ghrelin and anabolic/catabolic factors. Circulation. 2001;104:2034–8.
Cicoira M, Kalra P, Anker SD. Growth hormone resistance in chronic heart failure and its therapeutic implications. J Card Fail. 2003;9:219–26.
Doehner W, Rauchhaus M, Ponikowski P, Godsland IF, Von Haehling S, Okonko DO, et al. Impaired insulin sensitivity as an independent risk factor for mortality in patients with stable chronic heart failure. J Am Coll Cardiol. 2005;46:1019–26.
Kopple JD. Pathophysiology of protein-energy wasting in chronic renal failure. J Nutr. 1999;129:247S–51S.
Kalanthar-Zadeh K, Balakrishan VS. The kidney disease wasting: inflammation, oxidative stress, and diet–gene interaction. Hemodial Int. 2006;10:315–25.
Nomenclature consensus conference: kidney disease wasting. In: Fouque D, Kalantar-Zadeh K, editors. Organizing the XIII International Congress on Metabolism and Nutrition in Renal Disease, Merida, Yucatan, Mexico; 2006.
Kalantar-Zadeh K, Block G, McAllister CJ, Humphries MH, Kopple JD. Appetite and inflammation, nutrition, anemia, and clinical outcome in hemodialysis patients. Am J Clin Nutr. 2004;80:299–307.
Campbell KL, MacLaughlin HL. Unintentional weight loss is an independent predictor of mortality in a hemodialysis population. J Ren Nutr. 2010;20:414–8.
Johansen KL. Anabolic and catabolic mechanisms in end-stage renal disease. Adv Chronic Kidney Dis. 2009;16:501–10.
Mitch WE. Proteolytic mechanisms, not malnutrition, cause loss of muscle mass in kidney failure. J Ren Nutr. 2006;16:208–11.
Siew ED, Pupim LB, Majchrzak KM, Shintani A, Flakoll PJ, Ikizler TA. Insulin resistance is associated with skeletal muscle protein breakdown in non-diabetic chronic hemodialysis patients. Kidney Int. 2007;71:146–52.
Ikizler TA. Effects of glucose homeostasis on protein metabolism in patients with advanced chronic kidney disease. J Ren Nutr. 2007;17:13–6.
Guarnieri G, Zanetti M, Vinci P, Cattin MR, Barazzoni R. Insulin resistance in chronic uremia. J Ren Nutr. 2009;19:20–4.
Sandek A, Rauchhaus M, Anker SD, von Haehling S. The emerging role of the gut in chronic heart failure. Curr Opin Clin Nutr Metab Care. 2008;11:362–639.
Parks DA, Jacobson ED. Physiology of the splanchnic circulation. Arch Intern Med. 1985;145:1278–81.
Ding J, Cagnotti LJ, Huang Q, Xu DZ, Condon MR, Deitch EA. Hypoxia combined with Escherichia coli produces irreversible gut mucosal injury characterized by increased intestinal cytokine production and DNA degradation. Shock. 2001;16:189–95.
Ritz E. Intestinal–renal syndrome: mirage or reality? Blood Purif. 2011;31:70–6.
Mohi-ud-din K, Bali HK, Banerjee S, Sakhuja V, Jha V. Silent myocardial ischemia and high-grade ventricular arrhythmias in patients on maintenance hemodialysis. Ren Fail. 2005;27:171–5.
McIntyre CW. Haemodialysis-induced myocardial stunning in chronic kidney disease—a new aspect of cardiovascular disease. Blood Purif. 2010;29:105–10.
Wollert K, Drexler H. The role of interleukin-6 in the failing heart. Heart Fail Rev. 2001;6:95–103.
Ronco C, Cozzolino M. Mineral metabolism abnormalities and vitamin D receptor activation in cardiorenal syndromes. Heart Fail Rev; 2011 (in press).
Kanzaki M, Soda K, Gin PT, Kai T, Konishi F, Kawakami M. Erythropoietin attenuates cachectic events and decreases production of interleukin-6, a cachexia-inducing cytokine. Cytokine. 2005;32:234–9.
Perez-Fontan M, Cordido F, Rodriguez-Carmona A, Peteiro J, Garcia-Naveiro R, Garcia-Buela J. Plasma ghrelin levels in patients undergoing haemodialysis and peritoneal dialysis. Nephrol Dial Transplant. 2004;19:2095–100.
Yoshimoto A, Mori K, Sugawara A, et al. Associations between plasma ghrelin levels and body composition in end-stage renal disease: a longitudinal study. Plasma ghrelin and desacyl ghrelin concentrations in renal failure. J Am Soc Nephrol. 2002;13:2748–52.
Wren AM, Seal LJ, Cohen MA, et al. Ghrelin enhances appetite and increases food intake in humans. J Clin Endocrinol Metab. 2001;86:5992–5.
Wu R, Dong W, Cui X, et al. Ghrelin down-regulates proinflammatory cytokines in sepsis through activation of the vagus nerve. Ann Surg. 2007;245:480–6.
DeBoer MD, Zhu XX, Levasseur PR, et al. Ghrelin treatment causes increased food intake and retention of lean body mass in a rat model of cancer cachexia. Endocrinology. 2007;148:3004–12.
Baracos VE. Hypercatabolism and hypermetabolism in wasting states. Curr Opin Clin Nutr Metab Care. 2002;5:237–9.
Hedayat M, Mahmoudi MJ, Rose NR, Rezaei N. Proinflammatory cytokines in heart failure: double-edged swords. Heart Fail Rev. 2010;15:543–62.
von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7–8.
Acknowledgement
The authors of this manuscript certify that they comply with the Principles of Ethical Publishing in the Journal of Cachexia, Sarcopenia, and Muscle [104].
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Cicoira, M., Anker, S.D. & Ronco, C. Cardio-renal cachexia syndromes (CRCS): pathophysiological foundations of a vicious pathological circle. J Cachexia Sarcopenia Muscle 2, 135–142 (2011). https://doi.org/10.1007/s13539-011-0038-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13539-011-0038-2