
Abstract
Myoclonus can cause significant disability for patients. Myoclonus has a strikingly diverse array of underlying etiologies, clinical presentations, and pathophysiological mechanisms. Treatment of myoclonus is vital to improving the quality of life of patients with these disorders. The optimal treatment strategy for myoclonus is best determined based upon careful evaluation and consideration of the underlying etiology and neurophysiological classification. Electrophysiological testing including EEG (electroencephalogram) and EMG (electromyogram) data is helpful in determining the neurophysiological classification of myoclonus. The neurophysiological subtypes of myoclonus include cortical, cortical–subcortical, subcortical–nonsegmental, segmental, and peripheral. Levetiracetam, valproic acid, and clonazepam are often used to treat cortical myoclonus. In cortical–subcortical myoclonus, treatment of myoclonic seizures is prioritized, valproic acid being the mainstay of therapy. Subcortical–nonsegmental myoclonus may be treated with clonazepam, though numerous agents have been used depending on the etiology. Segmental and peripheral myoclonus are often resistant to treatment, but anticonvulsants and botulinum toxin injections may be of utility depending upon the case. Pharmacological treatments are often hampered by scarce evidence-based knowledge, adverse effects, and variable efficacy of medications.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myoclonus is defined as sudden, brief, lightning-like involuntary muscle jerks arising from the nervous system. Myoclonus may result from abrupt abnormal muscle activation or inhibition of muscle activation (negative myoclonus) [1]. Myoclonus can occur at rest, in response to stimulation, upon muscle activation, or any combination thereof. Such intrusions into normal movement disrupt activities of daily living, causing significant disability [2].
Myoclonus may occur in generalized, focal, multifocal, or segmental distributions. More than one distribution could be seen in the same patient or disease process. Identification of myoclonus can be challenging as other conditions cause brief jerking movements that may resemble myoclonus clinically. Such conditions could include dystonia, chorea, ballism, motor tics, and functional jerks [3]. Further, myoclonus may be subtle and can occur in repetitive fashion causing it to resemble other hyperkinetic movements such as tremor. It is critically important to correctly identify myoclonus as well as to avoid misidentification of myoclonus to ensure proper treatment.
There are many systems of classifying myoclonus including by etiology, examination findings, anatomical localization, and neurophysiological characteristics. This review will focus on using physiology-based classification to guide the treatment of myoclonus. The neurophysiological classification employed here is comprehensive and is designed to place similar physiology within the same category, allowing a uniform treatment approach of cases with the given electrophysiological characteristics. This approach to classification also presents the advantage of providing insights into the localization, underlying disease process, and responsiveness to certain treatments [4]. Understanding the underlying etiology of myoclonus also plays an important role in guiding treatment in tandem with electrophysiological data.
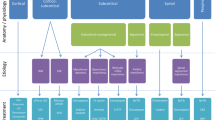
Regardless of the classification system that is being used, determining the optimal treatment of myoclonus is guided by the diagnostic work up and this begins with the clinical history and physical examination. Thorough diagnostic testing extends the history and physical examination to identify the etiology. This may include imaging of the brain and spinal cord as well as laboratory studies to assess for toxic, metabolic, infectious, inflammatory, paraneoplastic, degenerative, or genetic causes. An important goal of the clinical evaluation and classification of myoclonus is to identify whether a reversible cause exists. Electrophysiological testing is a valuable component of diagnostic testing, seeking both to confirm the presence of myoclonus and to determine the neurophysiological classification. Depending upon the clinical situation, the presence and etiology of myoclonus may be established at different junctures during the clinical evaluation. The neurophysiological classification can assist in determining the etiology of myoclonus in such cases in which an etiology is not apparent following initial diagnostic work up [5]. This diagnostic approach and resulting treatment strategy are summarized in Fig. 1.

The morphological differences in surface EMG morphology between a voluntary movement and a myoclonus discharge. On the left is the surface EMG recording of a healthy subject contracting the wrist flexors as quickly and as briefly as possible, demonstrating a burst duration of greater than 100 ms and a gradual build-up of EMG activity. On the right is a myoclonus discharge obtained from the same muscle group, demonstrating a brief duration of less than 50 ms and sharp rise time [11]
Pathophysiologic and Etiologic Classification of Myoclonus
There are numerous causes of myoclonus in terms of disease processes and pathophysiological mechanisms. Different etiologies may have a known favorable or unfavorable response to certain treatments. Additionally, knowledge of other potential comorbid features can influence treatment choice.
There are a number of different schemes used to classify the etiology of myoclonus. The most basic distinction involves determining “primary” myoclonus versus “secondary” or symptomatic myoclonus. Considering the immense variability in what could be considered a “primary” myoclonus, further distinctions may be helpful. Marsden et al. [1] proposed categories of physiologic, essential, and epileptic myoclonus in addition to symptomatic or secondary myoclonus. A modified version of this classification scheme is shown in Table 1. Physiologic myoclonus includes myoclonus that can occur in healthy humans. Examples can include hypnic jerks, fragmentary myoclonus, and startle responses. The frequency and intensity of physiologic myoclonus shows considerable variation among individuals. In the vast majority of cases, physiologic myoclonus does not require treatment. Identification of the normal nature of the phenomenon and reassurance of the patient may be all that is needed. Physiological forms of myoclonus can, however, become exaggerated or excessive and if significantly bothersome or debilitating, treatment could be warranted [6].
The category of essential myoclonus encompasses disorders of relatively isolated myoclonus that are minimally or nonprogressive and may cause varying degrees of functional disability. Included within this category are myoclonus–dystonia syndromes. Often genetically based, these disorders may be hereditary or sporadic. Mutations of the epsilon sarcoglycan gene (SGCE) are often implicated and display autosomal dominant inheritance [7]. The category of epileptic myoclonus includes seizure disorders with seizure semiology consisting of myoclonus. Myoclonus may occur as one of many components of seizure semiology, the sole manifestation of seizure, or may occur as one of multiple seizure types within an epilepsy syndrome. Symptomatic or secondary myoclonus encompasses a broad and most common category of myoclonus due to a variety of toxic, metabolic, infectious, inflammatory, degenerative, and structural causes [1].
Neurophysiological Classification of Myoclonus
Electrophysiological testing techniques that are useful in determining the neurophysiological classification of myoclonus include electroencephalogram (EEG) and multi-channel surface electromyogram (EMG), which are of most value when recorded concurrently. Even in situations in which access to EEG–EMG polygraphy may be limited, the recording of surface EMG can still be valuable. In addition to distinguishing myoclonus from other movement disorders, the electrophysiological characteristics of myoclonus discharges on surface EMG can provide important data as to the physiological classification. Myoclonus discharges recorded via EEG–EMG polygraphy can be analyzed using EEG–EMG back-averaging. Stimulus-induced EMG reflexes, corticomuscular coherence, and somatosensory evoked potentials are adjunctive studies that may assist in classifying myoclonus, particularly when a cortical origin is suspected [4, 8, 9].
Based upon the electrophysiological findings profile, myoclonus may be divided into the following categories: cortical, cortical–subcortical, subcortical–nonsegmental, segmental, and peripheral. Although not part of this classification scheme, functional or psychogenic jerks are an entity that may closely resemble myoclonus. Electrophysiological testing may give supportive evidence. The neurophysiological mechanisms, hence electrophysiological results of myoclonus, may vary across different etiologies of myoclonus. It is possible for more than one subtype of myoclonus to be present across one disease state and in the same individual [3, 4]. These nuances underscore the importance of properly classifying myoclonus by neurophysiology. The specific electrophysiological characteristics used to determine neurophysiological classification will be discussed separately for each myoclonus subtype.
Zutt et al. [5] studied the clinical utility of electrophysiological testing in classifying myoclonus according to anatomical localization. It was found that clinically suspected myoclonus was confirmed in 91% of cases. However, the subtype of myoclonus suspected prior to testing was often either changed or clarified by electrophysiological testing in over half of the cases. It was further demonstrated that an underlying etiology of myoclonus was subsequently determined in 53% of the patients in whom myoclonus was confirmed. Given that optimal treatment of myoclonus is heavily dependent upon properly diagnosing and classifying myoclonus, it follows that electrophysiological testing may aid in optimizing treatment of myoclonus.
General Treatment Strategy
When approaching treatment of myoclonus, the first consideration is whether a reversible or treatable cause exists. Examples include excising a spinal tumor causing propriospinal myoclonus, correcting a metabolic derangement, and withdrawing the offending drug, among others. This article is devoted to mostly symptomatic treatment. However, progress in defining new myoclonus etiologies is being made, which may in turn lead to advances in treatment. For example, the discovery of inflammatory syndromes that manifest myoclonus may provide an immune-based treatment approach for the myoclonus in some instances [10]. In many cases, treatment of the underlying cause of myoclonus is either not possible or provides insufficient relief of symptoms. In situations in which debilitating symptoms remain, symptomatic treatment is undertaken. For the rest of the article, “treatment” will refer to symptomatic treatment of myoclonus.
Due to the diverse etiologies and characteristics of myoclonus, the treatment of myoclonus is often challenging, and disability may remain after optimal symptomatic treatment occurs. Effective treatments across and even within different subtypes and etiologies may vary tremendously. Data are limited, especially with respect to double-blinded randomized controlled trials, and “off-label” treatments are often used. Polytherapy is often required for optimal symptom control. For these reasons, it is not possible to determine a precise algorithm for treating myoclonus. An individualized approach is often needed. Understanding comorbid features of the underlying disorder may assist in selecting an optimal treatment strategy. For example, selection of an agent known to treat seizures would be a reasonable choice for treating myoclonus that occurs in the setting of a seizure disorder. As will be outlined below in detail, different neurophysiological subtypes of myoclonus display differing responses to various treatments and therefore understanding the neurophysiological mechanism is crucial to determining the optimal treatment.
Cortical Myoclonus
Electrophysiological Testing Features
Cortical myoclonus is electrophysiologically characterized by brief surface EMG discharges typically less than 50 ms (Fig. 2). A pattern of agonist–antagonist cocontraction is often seen (Fig. 3). The distribution is often multifocal, though focal, segmental, and generalized distribution of myoclonus discharges can be seen. These discharges are often arrhythmic and may occur at rest, with stimulation, and/or with action. Cortical origin myoclonus is defined by a time-locked EEG cortical transient. This can be identified over the contralateral sensorimotor cortex. The latency between the EEG discharge and corresponding surface EMG discharge is between 6 and 22 ms when recorded from the upper extremity. Although such cortical transients are at times identified upon gross inspection of the EEG, back-averaging of EEG–EMG polygraphy is more reliably able to identify a corresponding cortical discharge (Fig. 4). When identifying myoclonus EMG discharges to include in back-averaging, it is recommended to have a motion sensor to assist with “jerk” confirmation [4].

The morphological differences in surface EMG morphology between a voluntary movement and a myoclonus discharge. On the left is the surface EMG recording of a healthy subject contracting the wrist flexors as quickly and as briefly as possible, demonstrating a burst duration of greater than 100 ms and a gradual build-up of EMG activity. On the right is a myoclonus discharge obtained from the same muscle group, demonstrating a brief duration of less than 50 ms and sharp rise time [11]

Surface EMG polygraphy is shown from the left upper extremity including the deltoids, biceps, triceps, wrist flexors, and wrist extensors. Myoclonus discharges are seen exhibiting cocontraction across flexor and extensor muscle groups as indicated by the arrows [3]
Cortical origin myoclonus may also be induced by a stimulus such as muscle stretch or sensory stimuli, so termed reflex myoclonus. This results from hyperexcitability of the sensorimotor integration. This stimulus sensitivity can be reflected electrophysiologically by finding enlarged cortical somatosensory evoked potentials, so termed “giant” SEPs. Single electrical stimulation of the median and/or tibial nerve can elicit myoclonus EMG discharges, serving as another means of demonstrating stimulus sensitivity. These enhanced long latency EMG reflexes, also termed C-reflexes at rest, can be recorded 40 to 60 ms following median nerve stimulation. The above findings demonstrating exaggerated cortical reflex phenomena can serve as supportive evidence of a cortical physiology of myoclonus [4, 8, 9].
There is considerable overlap between the physiological mechanisms of cortical myoclonus and focal motor seizures, both with presumed underlying hyperexcitability of the sensorimotor cortex. The specific locations and mechanisms of dysfunction within the pathways of the sensorimotor cortex remain unknown. It has been proposed that cortical myoclonus may exist as part of a spectrum including epilepsia partialis continua and focal motor seizures [12]. Cortical myoclonus has also been proposed to represent a fragment of focal epilepsy [13]). Focal motor seizures typically consist of paroxysmal repetitive focal surface EMG discharges of less than 100 ms that are confined to a focal distribution and demonstrate corresponding cortical transients that are either grossly visible on EEG or identified using EEG–EMG back-averaging (Fig. 4). Typically myoclonus associated with focal motor seizures will occur in paroxysms, but when occurring continuously at intervals no longer than every 10 s for periods of hours or longer, this is termed epilepsia partialis continua [13, 15].

Following EEG–EMG back-averaging, an averaged myoclonus discharge is pictured from the right wrist extensor along with the corresponding cortical transient at C3 [14]. The arrows indicate the onset of the positive deflection of the cortical transient and the onset of the myoclonus discharge occurring with a latency of ~ 20 ms
Treatment
Cortical myoclonus is often responsive to anti-epileptic treatment because these medications address the abnormal intrinsic cortical hyperexcitability that gives rise to cortical myoclonus.
Valproic acid has been found to be beneficial in cortical myoclonus. Valproic acid acts through multiple mechanisms, including increased synthesis of gamma-aminobutyric acid (GABA) in the nerve terminals and decreased degradation of GABA at the level of the synapse. Valproic acid also has some ability to decrease excitation by glutamate and alter ionic conductance [16]. Many patients need doses of 1200 to 2000 mg/day for myoclonus treatment [17]. The response of posthypoxic action myoclonus to valproic acid was reported in 1978 [18]. A series of eight Huntington disease patients with predominantly myoclonic movements showed a dose-dependent reduction in functional disability [19]. Valproic acid is often regarded as a first-line therapy in progressive myoclonic epilepsies [20].
Piracetam and related compounds including levetiracetam and brivaracetam have demonstrated efficacy in treating cortical myoclonus. These related compounds are believed to exert anti-myoclonic effect by binding to the synaptic vesicle protein 2A (SV2A), though the precise mechanism is unknown. Piracetam was shown to be superior to placebo for treating myoclonus in a double-blinded, randomized, and controlled trial of 21 patients with cortical myoclonus [21]. In an observational study of 40 patients with myoclonus of various etiologies, 16 improved clinically with the addition of piracetam, all of whom had cortical myoclonus. A double-blinded randomized crossover study of 20 patients with progressive myoclonic epilepsies demonstrated dose-dependent efficacy of piracetam [22]. An open-label clinical trial of 60 patients with myoclonus of different etiologies demonstrated effectiveness for treating myoclonus, particularly that of a cortical origin, both as monotherapy and polytherapy [23]. A wide variety of effective doses have been reported ranging from 2.4 to 24 g/day, emphasizing the importance of dosage titration. Possible side effects include gastrointestinal disturbance and hematologic abnormalities. Due to the high doses often required, the number and size of pills may limit medication adherence [24].
Levetiracetam, a related compound to piracetam, also acts on the SV2A receptor. Levetiracetam may exert anti-epileptic and anti-myoclonus effects through modulating calcium channels. Levetiracetam is widely used to treat seizure disorders and is generally well tolerated with few drug interactions, making this an attractive option for treating myoclonus. An open-label trial of 14 patients with electrophysiologically defined cortical myoclonus demonstrated evidence of clinical and electrophysiological improvement with levetiracetam polytherapy [25]. An open-label trial of 13 patients with myoclonic epilepsy demonstrated clinical improvement in 8 of the 13 patients when treated with levetiracetam [26]. Levetiracetam was reported to be of benefit in combination with clonazepam for treatment of myoclonic seizures in 17 patients with myoclonic epilepsy with ragged red fibers disease [27]. There have been reports of levetiracetam improving cortical myoclonus in patients with corticobasal degeneration and spinocerebellar ataxia [28,29,30]. Typical doses range from 1000 to 3000 mg daily. Brivaracetam is a derivative of levetiracetam, also acting on the SVA2 receptor with higher affinity compared with levetiracetam. Modulation of NMDA receptors and sodium currents may represent additional mechanisms of action. Brivaracetam has been FDA approved in recent years for the treatment of focal epilepsy. Brivaracetam has shown promise for treating posthypoxic cortical myoclonus in rodent models, though data in humans is lacking [31]. Brivaracetam has demonstrated efficacy for treating myoclonus in Unverricht–Lundborg disease and may also have fewer adverse effects than levitiracetam [32].
The possible mechanism of action in the case of clonazepam is facilitation of GABAergic transmission by affecting the benzodiazepine receptor and a decrease in 5-hydroxytryptophan utilization in the brain [33]. Clonazepam was reported as effective in treating myoclonus in a small series of three posthypoxic patients and two patients with presumed neurodegenerative disease [34]. The longer duration of action of clonazepam offers an advantage compared with some other benzodiazepine preparations. Relatively high doses of 7 to 18 mg daily have been used [35]. Clonazepam has been reported to benefit severe action myoclonus of multiple etiologies and may have some usefulness in polytherapy [17].
Perampanel is a selective noncompetitive α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid that the FDA approved in 2012 and currently used for treatment of focal and generalized epilepsy. A 2019 study suggested efficacy of low-dose perampanel in the treatment of cortical myoclonus in 18 patients with seizures and cortical myoclonus of varying etiologies. The mean dosage was 3.2 ± 2.1 mg/day. In addition to improvement myoclonus scales and measures of activities of daily living, treatment with perampanel resulted in increased latency and reduced amplitude of giant SEPs, suggesting that perampanel may reduce cortical hyperexcitability and hypersynchronization [3, 36]. There are multiple additional case reports of perampanel at dosages ranging from 4 to 10 mg improving cortical myoclonus of varying etiologies including myoclonic epilepsy, postsurgical, and posthypoxic myoclonus [36,37,38,39,40].
Other pharmacological treatments for cortical myoclonus have included other various anti-epileptic medications. The precise anti-myoclonus mechanism of these agents is unknown, but this likely relates to the suppression of excessive neuronal firing within the neuronal circuits that generate the cortical myoclonus. Some reports have documented the usefulness of lacosamide and agomelatine in treating posthypoxic myoclonus [41,42,43]. Zonisamide has demonstrated effectiveness as adjunctive therapy for treating myoclonus in patients with Unverricht–Lundborg disease and Lafora body disease [44, 45]. Progressive myoclonic epilepsies in particular often require polytherapy [13]. Sodium oxybate has demonstrated efficacy for the treatment of alcohol-responsive myoclonus in a rater blinded trial including patients with posthypoxic myoclonus and progressive myoclonic epilepsy [46]. Primidone and phenobarbital have also been used as adjunctive therapy in treating cortical action myoclonus [17]. Anti-epileptic medications that are not typically useful in treating cortical myoclonus include carbamazepine and phenytoin, which may in fact worsen myoclonus. The utility of these medications in treating myoclonus may be restricted to that which is associated with focal motor seizures [3].
Deep brain stimulation surgery has been performed in refractory cases of posthypoxic myoclonus with two cases demonstrating significant improvement in myoclonus severity scales after bilateral globus pallidus interna implantation [47, 48]. One case reports significant improvement in posthypoxic myoclonus due to perinatal anoxic injury following deep brain stimulation of the bilateral ventral intermediate nucleus [49].There have also been reports of deep brain stimulation showing efficacy in progressive myoclonic epilepsy. Targeting of the subthalamic nucleus has shown reduction in the frequency of myoclonic seizures. Implantation of the border zone between the subthalamic nucleus and substantia nigra pars reticularis has produced a beneficial effect upon cortical myoclonus [50,51,52]. A single case report demonstrated improvement of myoclonus with stimulation of the substantia nigra pars reticularis over a follow-up period of 24 months [53]. Although some positive results have been attained for deep brain stimulation in cases of cortical myoclonus, more study is needed before a consistent recommendation can be made.
Transcranial magnetic stimulation (TMS) has been used to study the balance between intracortical inhibition and excitation of motor pathways as well as the sensorimotor neuronal plasticity in different forms of myoclonus including progressive myoclonic epilepsy, myoclonus dystonia syndrome, and symptomatic myoclonus. TMS may also present potential for providing symptomatic treatment of myoclonus by restoring abnormal intracortical inhibition. It is thought that repetitive transcranial magnetic stimulation delivered at a low frequency (< 5 Hz) may enhance cortical inhibition [54]. A 2018 series of 9 patients with progressive myoclonic epilepsy underwent low-frequency repetitive TMS. One patient discontinued treatment due to worsening of myoclonus. The 8 patients who completed the protocol demonstrated a 25% improvement in myoclonus severity scales [55]. A series of 7 patients with epilepsia partialis continua of varying etiology were treated with low-frequency repetitive TMS with or without high-frequency repetitive TMS over the primary motor cortex. There was sustained benefit of greater than 1 day in 2 patients, no response in 2 patients, and the remaining 3 patients had benefit lasting 20 to 30 min [56]. A single case reported a longer duration benefit of 2 months following treatment with low-frequency repetitive TMS over the primary motor cortex for epilepsia partialis continua due to a cortical dysplasia [57]. In a case of childhood-onset cortical myoclonus manifesting clinically as tremor, clinical and electrophysiological improvement was reported with low-frequency repetitive TMS over the premotor cortex [58]. Transcranial magnetic stimulation may present a noninvasive and nonpharmacological method of treating cortical myoclonus, but further study is needed to address long-term efficacy.
Cortical–Subcortical Myoclonus
Etiology and Electrophysiological Testing Profile
Cortical–subcortical myoclonus most commonly arises in generalized epilepsy syndromes. Juvenile myoclonic epilepsy (JME) is the most common generalized epilepsy syndrome in which cortical–subcortical myoclonus is seen. It is important to note that all myoclonic epilepsies are not of cortical–subcortical myoclonus physiology. Progressive myoclonic epilepsies for example show cortical localization and physiology as discussed above. The seizures seen in generalized epilepsy syndromes arise from abnormal oscillations in bidirectional connections between cortical and subcortical structures that occur on a paroxysmal basis giving rise to generalized/bilateral myoclonus. Anterior thalamocortical circuitry has been implicated specifically in juvenile myoclonic epilepsy and in generalized absence seizures [59, 60]. Cortical–subcortical myoclonus usually occurs in paroxysms from rest and may occur in isolation or may clinically manifest as generalized seizures with a myoclonic component. The area of involvement of a single cortical–subcortical myoclonus discharge is much more diffuse and bilateral when compared with cortical myoclonus, owing to the bidirectional cortical and subcortical neuronal excitation that takes place. This excitatory discharge produces myoclonus when the discharge spreads to the primary motor cortices. For this reason, the clinical myoclonus seen in association with cortical–subcortical physiology is often bilateral and synchronous. Other symptoms such as loss of awareness may occur in conjunction with myoclonus when these excitatory discharges spread beyond the motor cortices to affect other structures [2,3,4].
Electrophysiologically, the prototypical corresponding findings are spike and/or polyspike activity with an aftercoming slow wave demonstrated on scalp EEG. Surface EMG myoclonus discharges are typically brief, often 50 to 100 ms, similar in duration to cortical myoclonus but potentially slightly longer. These surface EMG discharges occur in time-locked to fashion to a spike or polyspike and wave discharge on EEG. Enlarged cortical somatosensory evoked potentials have been reported in association with cortical–subcortical myoclonus, but are not consistently seen [4, 45]. Although there is evidence of cortical irritability in cortical–subcortical myoclonus, the differing clinical phenomenology and electrophysiological findings support a different pathological mechanism from purely cortical myoclonus. These important differences underscore the importance of treating these varieties of myoclonus with the electrophysiological mechanism in mind.
Treatment
Treatment most often consists of anti-epileptic medication with a primary goal of alleviating seizures. There are syndrome-specific treatment guidelines and algorithms that are often used in generalized epilepsy syndrome. Compared with the treatment of cortical myoclonus, there is some overlap, but a distinct profile of therapeutic efficacy is seen, largely owing to the differing neurophysiological generators. Evidence is limited with respect to the effect of anti-epileptic medications on myoclonus in these epilepsy syndromes. Valproic acid has been used as a first-line therapy for juvenile myoclonic epilepsy due to efficacy in terms of controlling seizures. The therapeutic effect of valproic acid in cortical–subcortical myoclonus may relate to the modulation of sodium and calcium ionic conductance. Lamotrigine has also been used to treat juvenile myoclonic epilepsy with differing results as to how this compares with valproic acid treatment. Initial data suggested superiority of valproic acid; however, subsequent studies have suggested slight superiority of lamotrigine. Notably, there were fewer adverse effects and events with use of lamotrigine as compared with valproic acid [61, 62]. There have also been rare reports of worsening myoclonus in juvenile myoclonic epilepsy treated with lamotrigine [63].
Levetiracetam and topiramate have also been reported to show efficacy in treating myoclonic seizures in JME [64]. The treatment of childhood absence epilepsy is typically aimed at controlling absence seizures. Ethosuximide and valproic acid are commonly used as a first-line treatment in childhood absence epilepsy. Double-blinded randomized controlled trial data have demonstrated equivalent efficacy of valproic acid and ethosuximide in treating childhood absence epilepsy with fewer adverse effects reported with ethosuximide. Data in myoclonic variants of childhood absence epilepsy are more limited, but suggest efficacy of valproic acid as well as valproic acid and ethosuximide cotherapy. Lamotrigine has been explored as a treatment of myoclonic absences, suggesting potential efficacy, but with very limited data [65, 66]. Intravenous valproic acid and levetiracetam may be useful in treating myoclonic status epilepticus [67]. Limited data exists showing efficacy of deep brain stimulation of the centromedian thalamic nucleus in treating treatment refractory generalized epilepsy; however, deep brain stimulation has not been studied specifically with respect to treatment of cortical–subcortical myoclonus [68].
Subcortical–Nonsegmental Myoclonus
Subcortical–nonsegmental myoclonus is a more heterogeneous entity in terms of physiological mechanisms and electrophysiological testing characteristics as compared with cortical myoclonus. There are numerous different subcortical locations from which myoclonus may arise yielding a wide variety of neuronal circuits through which myoclonic discharges may be transmit to descending motor pathways. On electrophysiological testing, the discharges of this myoclonus are often longer in duration compared with cortical myoclonus, as long as 200 ms, though this is not invariably the case. Varying patterns of muscle activation may be observed, including recruitment in a simultaneous rostral–caudal distribution, a purely descending caudal distribution, or in a multifocal distribution. Findings of cortical myoclonus such as EEG sharp waves, enlarged somatosensory evoked potentials, and enhanced long latency reflexes are not seen in subcortical myoclonus due to the lack of a cortical generator of the myoclonus [69]. Although it is important to note that these features are not seen, subcortical myoclonus should not be identified solely based upon the absence of these cortical features, as the number of technical and physiological factors may influence the recording of these features. Owing to the heterogeneity of mechanisms underlying subcortical myoclonus, a wide variety of treatments have been found to demonstrate partial efficacy. Anti-seizure medications used in cortical myoclonus such as valproic acid and levetiracetam are typically of limited utility in treating subcortical myoclonus [3].
Myoclonus–Dystonia Syndrome
A frequently cited example of subcortical–nonsegmental myoclonus includes myoclonus-dystonia syndrome, in which varying amounts of the myoclonus occur with or without dystonia. This is also termed hereditary essential myoclonus. One striking feature that has been seen in these disorders is a robust response to ethanol. Abnormal basal ganglia and cerebello-thalamic circuits through a genetic mechanism are believed underlie myoclonus–dystonia syndrome. The presence of GABAergic pathways within the basal ganglia may explain in part the therapeutic response to ethanol. A randomized double-blinded crossover trial of 23 patients with a myoclonus–dystonia syndrome demonstrated efficacy of zonisamide in treating myoclonus [70]. Clonazepam is anecdotally reported to have some efficacy in myoclonus dystonia syndromes, though clinical trial data is lacking. A single case report of alcohol-responsive myoclonus to gamma-hydroxybutyric acid has been reported [71]. Sodium oxybate, which is a sodium salt of gamma-hydroxybutyric acid, has been found to be effective in a single-blinded clinical trial, which included 3 patients with myoclonus–dystonia syndromes [46]. As previously described above in discussion of sodium oxybate use for cortical forms of myoclonus, this medication can have serious side effects including respiratory suppression and therefore should be approached cautiously. The lack of a robust response of GABA modulating medications matching that of ethanol suggests involvement of additional neurochemical pathways. Disruptions in serotonergic, dopaminergic, and cholinergic pathways may also play a role and present potential therapeutic mechanisms. Tetrabenazine was reported to effectively treat myoclonus in two patients with a myoclonus–dystonia syndrome [72].
Deep brain stimulation has been reported as effective for treatment of myoclonus dystonia syndromes using a globus pallidus interna (Gpi) [73] or Vim thalamus target [74]. A systematic review identifying 40 cases of myoclonus dystonia syndrome treated with deep brain stimulation concluded that 93.5% of patients had a response of 50% or greater on myoclonus severity scales with a mean improvement of 72.6% over a mean follow-up period of 27/2 months. The improvement was similar for both Gpi and Vim in terms of myoclonus, but dystonia improved more with the Gpi target and fewer adverse events were reported [75]. Included in the above review is a case series of 10 patients with myoclonus dystonia who underwent bilateral Gpi and Vim deep brain stimulation. This showed improvement in myoclonus severity for both targets when stimulated separately, but more adverse effects were seen with Vim stimulation. In 6 patients, there was slightly better efficacy with stimulation of both the Gpi and Vim concurrently. Although larger scale studies are needed to validate these results, Gpi deep brain stimulation appears promising for treatment of myoclonus dystonia syndrome and a Vim target may provide an additional option for patients who may be refractory to Vim stimulation [76].
Opsoclonus–Myoclonus Syndrome
Opsoclonus–myoclonus syndrome is an example of subcortical–nonsegmental myoclonus believed to arise from brainstem pathways. Although classically attributed to a paraneoplastic syndrome related to pediatric neuroblastoma, a variety of inflammatory, infectious, and structural causes can underlie this syndrome, especially in adults. In the case of a paraneoplastic etiology, identifying and addressing the associated malignancy becomes the priority. Inflammatory or autoimmune causes as expected may be addressed using immunotherapy. Aside from treating the underlying cause, clonazepam has been reported as effective in some cases of opsoclonus–myoclonus syndrome. Other agents reported as effective in case reports include valproic acid, piracetam, topiramate, and reserpine [3, 77,78,79].
Reticular Reflex Myoclonus
Reticular reflex myoclonus is a rare entity that has been reported in the setting of uremia and recovery from hypoxia. Myoclonus occurs in a generalized distribution, predominantly in the proximal upper extremity and truncal muscles and may be induced by muscle activation or startle. There may or may not be accompanying generalized discharges seen on EEG. The relationship to seizure is therefore uncertain. The surface EMG may demonstrate sequential activation of muscles innervated by brainstem nuclei. Typical findings of cortical myoclonus such as a focal cortical sharp wave and enlarged somatosensory evoked potentials are not seen. Intravenous clonazepam and 5-hydroxytryptophan have previously been reported as effective in case reports, though neither is commonly used presently. Reticular reflex myoclonus in the setting of hypoxia or uremia is often reversible with correction of the underlying metabolic derangement [79, 80].
Propriospinal Myoclonus
Propriospinal myoclonus is a form of nonsegmental myoclonus resulting in jerking of truncal muscles from a spinal cord generator. The electrophysiological signature of propriospinal myoclonus is surface EMG discharges arising from a particular level of the spinal cord with concurrent, contiguous activation of rostral and caudal muscle groups. This may involve the upper and/or lower extremity muscles. The pattern of activation is typically bilateral and symmetric. The duration of surface EMG discharges may be variable in length and may be longer than in cortical or cortical–subcortical myoclonus, possibly lasting in excess of 300 ms. Propriospinal myoclonus is often refractory to pharmacological treatment. Clonazepam has been the most consistently reported to be effective. Zonisamide has been reported as effective, albeit in a minority of cases. A variety of other pharmacological treatments have been reported as mildly effective in isolated cases including piracetam, levetiracetam, valproic acid, oxcarbazepine, carbamazepine, and baclofen. Propriospinal myoclonus can be caused by irritative or compressive lesions affecting the spinal cord. There are reports of alleviation of propriospinal myoclonus following surgical removal of a causative structural lesion. Assessment for such a structural lesion with spinal cord imaging is therefore important in proposing appropriate treatment [79, 81, 82].
It is essential to maintain awareness that functional jerks may closely resemble propriospinal myoclonus both in terms of clinical and electrophysiological characteristics. Studies have shown that the expected electrophysiological pattern expected in propriospinal myoclonus can be seen in the setting of functional truncal jerks and can also be effectively mimicked by healthy volunteers [83,84,85]. The optimal treatment of functional jerks differs from that of propriospinal myoclonus (see the section on functional myoclonus below).
Segmental Myoclonus
Electrophysiological Testing Profile
Segmental myoclonus presents with distinct neurophysiological features from subcortical–nonsegmental myoclonus. Segmental myoclonus arises from a generator at a particular segment or contiguous segments usually of the brainstem or spinal cord. The surface EMG therefore shows synchronous activation of muscles innervated by involved segments of the nervous system. Spinal segmental myoclonus is distinguished from other forms of myoclonus by the involvement of muscles corresponding to 1 to 3 contiguous spinal levels. Segmental myoclonus in contrast to peripheral myoclonus may also occur bilaterally. The duration of surface EMG discharges may vary considerably, lasting anywhere from 50 ms up to as long as 500 ms. The myoclonus discharges are often rhythmic with frequencies varying from 0.2 to 8 Hz. Segmental myoclonus is typically persistent and unaffected by state of consciousness, motor activity, or sensory stimuli. The EEG and somatosensory evoked potentials are generally normal. However, brainstem auditory evoked potentials have been reported to be abnormal in some cases of palatal myoclonus. Peripheral nerve stimulation in spinal segmental myoclonus has been reported evoke EMG discharges in affected muscles at latencies in excess of 40 ms [4, 86, 87].
Palatal Myoclonus
The most common example of segmental myoclonus is palatal myoclonus. Palatal myoclonus may be symptomatic, arising as a result of lesions of the dentato-rubro-olivary pathway (also termed Guillain Mollaret triangle or myoclonic triangle), in which case other symptoms or findings localizable to the brainstem or cerebellum may be seen. Essential or idiopathic palatal myoclonus may also occur, in which case the palatal myoclonus may be an isolated finding. Symptoms of bothersome clicking in the ear may be reported. Palatal myoclonus is often resistant to treatment and there is no agent that has shown consistent benefit. There are reports of numerous different medications showing efficacy in small numbers of cases. Medications reported as beneficial include clonazepam, baclofen [86], sumatriptan [88], lamotrigine [89], trihexyphenidyl, carbamazepine, and piracetam [90]. Botulinum toxin injections have also shown promise in alleviating palatal myoclonus [91,92,93]. A 2014 case series of patients with palatal myoclonus who underwent botulinum toxin injections documents improvement in symptom of 12 of 14 patients. A previous 2005 review demonstrated similar findings with 4 of 5 patients showing symptomatic improvement after botulinum toxin injections [93, 94]. Surgical intervention such as tensor veli palatini tenotomy and occlusion of the Eustachian tube have been used to treat severe, disabling ear clicking resulting from treatment refractory palatal myoclonus [95]. It is important to consider the possibility of a functional disorder, as there have been reported cases of functional palatal tremor reported [96,97,98].
Spinal Segmental Myoclonus
Numerous etiologies may cause segmental myoclonus including structural lesions (vascular, neoplastic, inflammatory, infectious, etc.) as well as idiopathic causes [4, 11]. The pathophysiological generator is believed to be a loss of inhibition of spinal interneurons leading to hyperexcitability of anterior horn cells within the affected segment [87]. Spinal segmental myoclonus, similar to palatal myoclonus, often proves refractory to medical therapy and thus a wide variety of medications have been reported as useful in small numbers of cases. The most consistently useful medication has been clonazepam, others including tetrabenazine, trihexyphenidyl, baclofen, carbamazepine, piracetam, and levetiracetam [99, 100]. Given the localized distribution of spinal segmental myoclonus, botulinum toxin injections may be beneficial both in terms of treating involuntary movement and associated discomfort. There are case reports of botulinum toxin injections successfully treating spinal segmental myoclonus affecting the neck, shoulder, and lower extremity [101,102,103]. Spinal segmental myoclonus may be due to a variety of causes, some of which are attributable to infectious or inflammatory conditions that could spontaneously change or resolve over time. Therefore, care must be taken not to erroneously attribute treatment effect in such a scenario [3].
Peripheral Myoclonus
Peripheral myoclonus often occurs as localized myoclonic jerks that arise from a peripheral nervous system generator. The surface EMG demonstrates discharges of varying length arising from the same peripheral nerve distribution. Within a single nerve distribution, multiple muscles may be affected and these muscles would be expected to show similar electrophysiological firing patterns. The myoclonus may occur intermittently with discharges occurring in a semi-rhythmic or rhythmic pattern. As expected, the findings associated with cortical myoclonus such as a cortical transient or enlarged somatosensory evoked potentials are not seen. Proposed mechanisms of peripheral myoclonus include direct motor nerve irritation due to compression or alteration of central nervous system motor pathways secondary to a peripheral nerve lesion. The most common example of peripheral myoclonus is hemifacial spasm, which typically occurs in association with vascular compression of unilateral cranial nerve VII.
Peripheral myoclonus is often treatable by botulinum toxin injections due to the focal distribution of musculature involved. Hemifacial spasm is known to respond well to botulinum toxin injections, demonstrating subjective improvement of 80% in ~ 80% of patients [104]. Oral medications are best used as adjunctive therapy for hemifacial spasm, as complete relief of symptoms is not typical. Carbamazepine has shown sustained improvement in 35% of patients with complete relief occurring even less often [105]. Gabapentin has also shown efficacy, with an open-label trial of 23 patients demonstrating at least 70% improvement in 69% of patients [106]. Surgical treatment is a consideration in the presence of a compressive lesion. Favorable results have been seen in up to 90% of patients who undergo microvascular decompression for hemifacial spasm, though symptoms may recur following initial success [107, 108].
Functional Jerks
An entity distinct from myoclonus, yet important to recognize in the differential diagnosis of myoclonus, is that of functional jerks. Functional movement disorders may closely resemble myoclonus clinically. However, the more prolonged duration of functional jerks may be detected visually by the clinician. Electrophysiological testing can be helpful as supportive evidence in differentiating functional jerks from true myoclonus with functional movements demonstrating longer duration of surface EMG discharges, usually greater than 100 ms as well as marked variability of duration and distribution. A nonstereotyped pattern of muscle recruitment may also be seen, though an agonist–antagonist pattern of muscle activation typical of myoclonus can be seen in functional jerks as well. In stimulus-induced jerks, a response latency of greater than 100 ms suggests a functional etiology [109].
Importantly, it has been recognized that in some cases, psychogenic movements may resemble myoclonus even electrophysiologically for those types of myoclonus with longer EMG discharge duration. This has especially been described in the case of functional truncal jerks that closely resemble propriospinal myoclonus. Some cases of palatal myoclonus have been identified to be functional in etiology [96,97,98]. The history and physical examination should carefully evaluate for risk factors or other features that may suggest a functional etiology. When diagnosing a functional movement disorder, it is also important to recognize that a functional disorder may co-exist with an organic movement disorder. For this reason, care must be taken to adequately evaluate for such a disorder.
Consideration may also be given to performing EEG–EMG back-averaging for the purpose of identifying a Bereitschaftspotential [84]. A Bereitschaftspotential is a negative EEG potential reflecting activity of premotor areas that occurs before a voluntary movement. The early phase of the Bereitschaftspotential begins 1 to 2 s before a voluntary movement and demonstrates a slowly rising negative polarity that is widespread in distribution but maximal at the vertex. The late phase of the Bereitschaftspotential begins at 450 to 650 ms before a voluntary movement at which point the negative shift becomes steeper and more lateralized contralateral to the movement. This may be a useful technique to evaluation of functional myoclonus; however, the identification of a Bereitschaftspotential may be technically challenging. For this reason, the absence of a Bereitschaftspotential should not be used as evidence to exclude a functional etiology [4, 109]. Event-related desynchronization refers to the reduction of beta and gamma oscillations that occur prior to either self-paced or cued voluntary movement, which may reflect attention directed at motor control. This may also serve as a useful electrophysiological marker of functional movements [110].
Once a functional movement disorder has been diagnosed, the first step in treatment is explaining the functional nature of symptoms to the patient. A multidisciplinary treatment approach is often needed, which should be individualized to the patient. Medication treatment is generally not indicated for functional movement disorders, with the exception of appropriate pharmacological management of psychiatric comorbidities. Successful treatment modalities have included cognitive behavioral therapy, biofeedback, physical therapy, physical activity, transcutaneous electrical nerve stimulation (TENS), and transcranial magnetic stimulation [111, 112].
Conclusion
Myoclonus of all varieties can cause significant disability. Treatment of myoclonus is vital to improving the quality of life of patients with these disorders. Owing to the diverse etiologies and classifications of myoclonus, a wide array of potentially effective treatments are reported. Still, the treatment of myoclonus remains challenging in the face of a paucity of large randomized clinical trials, likely owing in large part to the rarity of these diverse etiologies of myoclonus. Pharmacological treatments are often hampered by side effects, variable efficacy, and a lack of evidence-based guidelines. Neuromodulation appears promising in select cases, but requires additional study. Determining the etiology of myoclonus as well as the neurophysiological classification are important steps in guiding selection of the optimal treatment regimen. Electrophysiological testing is of great value in this regard. Further study of the role of neurophysiological testing in guiding clinical management of myoclonus would be beneficial in understanding the optimal approach to treatment.
References
Marsden CD, Hallett M, Fahn S. The nosology and pathophysiology of myoclonus. In: Marsden CD, Fahn S, editors. Movement Disorders. London: Butterworths; 1983. pp. 196–248.
Caviness JN, Brown P. Myoclonus: current concepts and recent advances. Lancet Neurol. 2004;3:598–607.
Caviness JN. Treatment of myoclonus. Neurotherapeutics. 2014;11(1):188–200.
Caviness JN Clinical neurophysiology of myoclonus. In: Hallett M, editor. Movement Disorders. Handbook of Clinical Neurophysiology. Amsterdam: Elsevier; 2003. pp. 521–548.
Zutt R, Elting JW, van Zijl JC, et al. Electrophysiologic testing aids diagnosis and subty** of myoclonus. Neurology. 2018;90(8):e647–e657.
Vetrugno R, Plazzi G, Provini F, Liguori R, Lugaresi E, Montagna P. Excessive fragmentary hypnic myoclonus: clinical and neurophysiological findings. Sleep Med. 2002;3(1):73–76.
Kinugawa K, Vidailhet M, Clot F, Apartis E, Grabli D, Roze E. Myoclonus-dystonia: an update. Mov Disord. 2009;24(4):479–489.
Chen KS, Chen R. Principles of electrophysiological assessments for movement disorders. J Mov Disord. 2020;13(1):27-38. doi:https://doi.org/10.14802/jmd.19064
Shibasaki H. Electrophysiological studies of myoclonus. Muscle Nerve. 2000;23(3):321-335. doi:https://doi.org/10.1002/(sici)1097-4598(200003)23:3<321::aid-mus3>3.0.co;2-3
Baizabal-Carvallo JF, Jankovic J. Autoimmune and paraneoplastic movement disorders: an update. J Neurol Sci. 2018;385:175-184. doi:https://doi.org/10.1016/j.jns.2017.12.035
Caviness JN. Myoclonus. Mayo Clin Proc. 1996;71(7):679–688.
Obeso JA, Rothwell JC, Marsden CD. The spectrum of cortical myoclonus. From focal reflex jerks to spontaneous motor epilepsy. Brain. 1985;108 (Pt 1). doi:https://doi.org/10.1093/brain/108.1.193
Hallett M. Myoclonus: relation to epilepsy. Epilepsia. 1985;26 Suppl 1:S67-S77. doi:https://doi.org/10.1111/j.1528-1157.1985.tb05726.x
Neuroscan [computer software]. Version 4.3. El Paso, TX: Compumedics, 2003.
Thomas JE, Reagan TJ, Klass DW. Epilepsia partialis continua. A review of 32 cases. Arch Neurol. 1977;34(5):266-275. doi:https://doi.org/10.1001/archneur.1977.00500170020003
VanDongen AM, VanErp MG, Voskuyl RA. Valproate reduces excitability by blockage of sodium and potassium conductance. Epilepsia. 1986;27(3):177–182.
Obeso JA, Artieda J, Rothwell JC, Day B, Thompson P, Marsden CD. The treatment of severe action myoclonus. Brain. 1989;112 ( Pt 3):765–777.
Fahn S. Posthypoxic action myoclonus: review of the literature and report of two new cases with response to valproate and estrogen. Adv Neurol. 1979;26:49–84.
Saft C, Lauter T, Kraus PH, Przuntek H, Andrich JE. Dose-dependent improvement of myoclonic hyperkinesia due to valproic acid in eight Huntington’s disease patients: a case series. BMC Neurol. 2006;6:11. Published 2006 Feb 28.
Ferlazzo E, Trenite DK, Haan GJ, et al. Update on pharmacological treatment of progressive myoclonus epilepsies. Curr Pharm Des. 2017;23(37):5662–5666.
Brown P, Steiger MJ, Thompson PD, et al. Effectiveness of piracetam in cortical myoclonus. Mov Disord. 1993;8(1):63–68.
Koskiniemi M, Van Vleymen B, Hakamies L, Lamusuo S, Taalas J. Piracetam relieves symptoms in progressive myoclonus epilepsy: a multicentre, randomised, double blind, crossover study comparing the efficacy and safety of three dosages of oral piracetam with placebo. J Neurol Neurosurg Psychiatry. 1998;64(3):344–348.
Ikeda A, Shibasaki H, Tashiro K, Mizuno Y, Kimura J. Clinical trial of piracetam in patients with myoclonus: nationwide multiinstitution study in Japan. The Myoclonus/Piracetam Study Group. Mov Disord. 1996;11(6):691–700.
Michelucci R, Pasini E, Riguzzi P, Andermann E, Kälviäinen R, Genton P. Myoclonus and seizures in progressive myoclonus epilepsies: pharmacology and therapeutic trials. Epileptic Disord. 2016;18(S2):145–153.
Striano P, Manganelli F, Boccella P, Perretti A, Striano S. Levetiracetam in patients with cortical myoclonus: a clinical and electrophysiological study. Mov Disord. 2005;20(12):1610–1614.
Magaudda A, Gelisse P, Genton P. Antimyoclonic effect of levetiracetam in 13 patients with Unverricht-Lundborg disease: clinical observations. Epilepsia. 2004;45(6):678–681.
Su LJ, Wang YL, Han T, et al. Antimyoclonic effect of levetiracetam and clonazepam combined treatment on myoclonic epilepsy with ragged-red fiber syndrome with m.8344A>G mutation. Chin Med J (Engl). 2018;131(20):2433–2438.
Cho JW, Lee JH. Suppression of myoclonus in corticobasal de-generation by levetiracetam. J Mov Disord. 2014;7(1):28–30.
Kovács T, Farsang M, Vitaszil E, et al. Levetiracetam reduces myoclonus in corticobasal degeneration: report of two cases. J Neural Transm (Vienna). 2009;116(12):1631–1634.
Orsucci D, Ienco EC, Rocchi A et al. Levetiracetam-responsive myoclonus in spinocerebellar ataxia type 15. Mov Disord. 2013;28(10):1465.
Tai KK, Truong DD. Brivaracetam is superior to levetiracetam in a rat model of post-hypoxic myoclonus. J Neural Transm (Vienna). 2007;114(12):1547–1551.
Kälviäinen R, Genton P, Andermann E, et al. Brivaracetam in Unverricht-Lundborg disease (EPM1): results from two randomized, double-blind, placebo-controlled studies. Epilepsia. 2016;57(2):210–221.
Jenner P, Pratt JA, Marsden CD. Mechanism of action of clonazepam in myoclonus in relation to effects on GABA and 5-HT. Adv Neurol. 1986;43:629–643.
Goldberb MA, Dorman JD. Intention myoclonus: successful treatment with clonazepam. Neurology. 1976;26(1):24–6.
Fahn S. Posthypoxic action myoclonus: literature review update. Adv Neurol. 1986;43:157–169.
Oi K, Neshige S, Hitomi T, et al. Low-dose perampanel improves refractory cortical myoclonus by the dispersed and suppressed paroxysmal depolarization shifts in the sensorimotor cortex. Clin Neurophysiol. 2019;130(10):1804–1812.
Iijima M, Oguni H, Kobayashi M, Kitagawa K. Perampanel improved intractable myoclonus in two patients with myoclonus epilepsy. eNeurologicalSci. 2019;17:100215. Published 2019 Nov 18. doi:https://doi.org/10.1016/j.ensci.2019.100215
Santamarina E, Sueiras M, Lidón RM, et al. Use of perampanel in one case of super-refractory hypoxic myoclonic status: case report. Epilepsy Behav Case Rep. 2015;4:56-59. Published 2015 Aug 8. doi:https://doi.org/10.1016/j.ebcr.2015.06.007
Steinhoff BJ, Bacher M, Kurth C, Staack AM, Kornmeier R. Add-on perampanel in Lance-Adams syndrome. Epilepsy Behav Case Rep. 2016;6:28-29. Published 2016 Jun 1. doi:https://doi.org/10.1016/j.ebcr.2016.05.001
Bianchini S, Bellantoni G, Albergati A, Magrassi L. Postsurgical cortical myoclonus responsive to perampanel. Neurol Clin Pract. 2018;8(2):159-161. doi:https://doi.org/10.1212/CPJ.0000000000000440
de la González Aleja J, Saiz-Díaz RA, De la Peña P. Relief of intractable posthypoxic myoclonus after administration of agomelatine. Clin Neuropharmacol. 2012;35:258–259.
Galldiks N, Timmermann L, Fink GR, Burghaus L. Posthypoxic myoclonus (Lance-Adams syndrome) treated with lacosamide. Clin Neuropharmacol. 2010;33:216–217.
Gupta HV, Caviness JN. Post-hypoxic myoclonus: current concepts, neurophysiology, and treatment. Tremor Other Hyperkinet Mov (N Y). 2016;6:409.
Kyllerman M, Ben-Menachem E. Zonisamide for progressive myoclonus epilepsy: long-term observations in seven patients. Epilepsy Res. 1998;29(2):109–14.
Yoshimura I, Kaneko S, Yoshimura N, Murakami T. Long-term observations of two siblings with Lafora disease treated with zonisamide. Epilepsy Res. 2001;46(3):283–287.
Frucht SJ, Houghton WC, Bordelon Y, Greene PE, Louis ED. A single-blind, open-label trial of sodium oxybate for myoclonus and essential tremor. Neurology. 2005;65(12):1967–1969.
Asahi T, Kashiwazaki D, Dougu N, et al. Alleviation of myoclonus after bilateral pallidal deep brain stimulation for Lance-Adams syndrome. J Neurol. 2015;262:1581–1583.
Ramdhani RA, Frucht SJ, Kopell BH. Improvement of post-hypoxic myoclonus with bilateral pallidal deep brain stimulation: a case report and review of the literature. Tremor Other Hyperkinet Mov (N Y). 2017;7:461. Published 2017 May 19. doi:https://doi.org/10.7916/D8NZ8DXP
Kobayashi K, Katayama Y, Otaka T, et al. Thalamic deep brain stimulation for the treatment of action myoclonus caused by perinatal anoxia. Stereotact Funct Neurosurg. 2010;88(4):259-263. doi:https://doi.org/10.1159/000315464
Anderson DG, Németh AH, Fawcett KA, Sims D, Miller J, Krause A. Deep brain stimulation in three related cases of North Sea progressive myoclonic epilepsy from South Africa. Mov Disord Clin Pract. 2016;4(2):249–253. Published 2016 Jun 16.
Wille C, Steinhoff BJ, Altenmüller DM, et al. Chronic high-frequency deep-brain stimulation in progressive myoclonic epilepsy in adulthood--report of five cases. Epilepsia. 2011;52(3):489–496.
Vesper J, Steinhoff B, Rona S, et al. Chronic high-frequency deep brain stimulation of the STN/SNr for progressive myoclonic epilepsy. Epilepsia. 2007;48(10):1984-1989. doi:https://doi.org/10.1111/j.1528-1167.2007.01166.x
di Giacopo A, Baumann CR, Kurthen M, Capecchi F, Sürücü O, Imbach LL. Selective deep brain stimulation in the substantia nigra reduces myoclonus in progressive myoclonic epilepsy: a novel observation and short review of the literature. Epileptic Disord. 2019;21(3):283-288. doi:https://doi.org/10.1684/epd.2019.1072
Nardone R, Versace V, Höller Y, et al. Transcranial magnetic stimulation in myoclonus of different aetiologies. Brain Res Bull. 2018;140:258–269.
Rossi Sebastiano D, Magaudda A, Quartarone A, et al. Effect of repetitive transcranial magnetic stimulation on action myoclonus: a pilot study in patients with EPM1. Epilepsy Behav. 2018;80:33-36. doi:https://doi.org/10.1016/j.yebeh.2017.11.031
Rotenberg A, Bae EH, Takeoka M, Tormos JM, Schachter SC, Pascual-Leone A. Repetitive transcranial magnetic stimulation in the treatment of epilepsia partialis continua. Epilepsy Behav. 2009;14(1):253-257. doi:https://doi.org/10.1016/j.yebeh.2008.09.007
Misawa S, Kuwabara S, Shibuya K, Mamada K, Hattori T. Low-frequency transcranial magnetic stimulation for epilepsia partialis continua due to cortical dysplasia. J Neurol Sci. 2005;234(1-2):37-39. doi:https://doi.org/10.1016/j.jns.2005.03.035
Houdayer E, Devanne H, Tyvaert L, Defebvre L, Derambure P, Cassim F. Low frequency repetitive transcranial magnetic stimulation over premotor cortex can improve cortical tremor. Clin Neurophysiol. 2007;118(7):1557-1562. doi:https://doi.org/10.1016/j.clinph.2007.04.014
Snead OC. Basic mechanisms of generalized absence seizures. Ann Neurol. 1995;37:146–57.
Deppe M, Kellinghaus C, Duning T, et al. Nerve fiber impairment of anterior thalamocortical circuitry in juvenile myoclonic epilepsy. Neurology. 2008;71:1981–1985
Wallace SJ. Myoclonus and epilepsy in childhood: a review of treatment with valproate, ethosuximide, lamotrigine and zonisamide. Epilepsy Res. 1998;29(2):147–54.
Machado RA, García VF, Astencio AG, Cuartas VB. Efficacy and tolerability of lamotrigine in juvenile myoclonic epilepsy in adults: a prospective, unblinded randomized controlled trial. Seizure. 2013;22(10):846–855.
Carrazana EJ, Wheeler SD. Exacerbation of juvenile myoclonic epilepsy with lamotrigine. Neurology. 2001;56(10):1424–5.
Specchio LM, Gambardella A, Giallonardo AT, et al. Open label, long-term, pragmatic study on levetiracetam in the treatment of juvenile myoclonic epilepsy. Epilepsy Res. 2006;71(1):32–39.
Manganotti P, Tamburin S, Bongiovanni LG, Zanette G, Fiaschi A. Motor responses to afferent stimulation in juvenile myoclonic epilepsy. Epilepsia 2004;45:77–80.
Glauser TA, Cnaan A, Shinnar S, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy: initial monotherapy outcomes at 12 months. Epilepsia. 2013;54(1):141–155.
Sheth RD, Gidal BE. Intravenous valproic acid for myoclonic status epilepticus. Neurology. 2000;54(5):1201.
Valentín A, García Navarrete E, Chelvarajah R, et al. Deep brain stimulation of the centromedian thalamic nucleus for the treatment of generalized and frontal epilepsies. Epilepsia. 2013;54(10):1823–1833.
Li JY, Cunic DI, Paradiso G, et al. Electrophysiological features of myoclonus-dystonia. Mov Disord. 2008;23(14):2055–2061.
Hanique E, Vidailhet M, Cozic N et al. A randomized, controlled, double-blind crossover trial of zonisamide in myoclonus-dystonia. Neurology 2016; 86:1729 – 1735.
Priori A, Bertolasi L, Pesenti A, Cappellari A, Barbieri S. Gamma-hydroxybutyric acid for alcohol-sensitive myoclonus with dystonia. Neurology. 2000;54(8):1706.
Luciano AY, **nah HA, Pfeiffer RF, Truong DD, Nance MA, LeDoux MS. Treatment of myoclonus-dystonia syndrome with tetrabenazine. Parkinsonism Relat Disord. 2014;20(12):1423–1426.
Roze E, Vidailhet M, Hubsch C, Navarro S, Grabli D. Pallidal stimulation for myoclonus-dystonia: ten years’ outcome in two patients. Mov Disord. 2015;30(6):871–872.
Kuncel AM, Turner DA, Ozelius LJ, Greene PE, Grill WM, Stacy MA. Myoclonus and tremor response to thalamic deep brain stimulation parameters in a patient with inherited myoclonus-dystonia syndrome. Clin Neurol Neurosurg. 2009;111(3):303–306.
Rughani AI, Lozano AM. Surgical treatment of myoclonus dystonia syndrome. Mov Disord. 2013;28(3):282-287. doi:https://doi.org/10.1002/mds.25326
Gruber D, Kühn AA, Schoenecker T, et al. Pallidal and thalamic deep brain stimulation in myoclonus-dystonia. Mov Disord. 2010;25(11):1733-1743. doi:https://doi.org/10.1002/mds.23312
Fernandes TD, Bazan R, Betting LE, da Rocha FC. Topiramate effect in opsoclonus-myoclonus-ataxia syndrome. Arch Neurol 2012; 69:133.
Bataller L, Graus F, Saiz A, Vilchez JJ, Spanish Opsoclonus-Myoclonus Study Group. Clinical outcome in adult onset idiopathic or paraneoplastic opsoclonus-myoclonus. Brain. 2001;124(Pt 2):437–443.
Mills K, Mari Z. An update and review of the treatment of myoclonus. Curr Neurol Neurosci Rep. 2015;15(1):512.
Chadwick D, French AT. Uraemic myoclonus: an example of reticular reflex myoclonus?. J Neurol Neurosurg Psychiatry. 1979;42(1):52–55.
Roze E, Bounolleau P, Ducreux D, et al. Propriospinal myoclonus revisited: clinical, neurophysiologic, and neuroradiologic findings. Neurology. 2009;72(15):1301–1309.
Antelmi E., Provini F. Propriospinal myoclonus: the spectrum of clinical and neurophysiological phenotypes. Sleep Med. Rev. 2015;22:54–63.
Kang S, Sohn Y. Electromyography patterns of propriospinal myoclonus can be mimicked voluntarily. Mov Disord 2006; 21: 1241– 1244.
van der Salm SM, Koelman JH, Henneke S, van Rootselaar AF, Tijssen MA. Axial jerks: a clinical spectrum ranging from propriospinal to psychogenic myoclonus. J Neurol. 2010;257(8):1349–1355.
Esposito M, Edwards MJ, Bhatia KP, Brown P, Cordivari C. Idiopathic spinal myoclonus: a clinical and neurophysiological assessment of a movement disorder of uncertain origin. Mov Disord. 2009;24(16):2344–2349.
Jankovic J, Pardo R. Segmental myoclonus. Clinical and pharmacologic study. Arch Neurol. 1986;43:1025–1031
Termsarasab P, Thammongkolchai T, Frucht SJ. Spinal-generated movement disorders: a clinical review [published correction appears in J Clin Mov Disord. 2016 22;3:18]. J Clin Mov Disord. 2015;2:18. Published 2015 Dec 24. doi:https://doi.org/10.1186/s40734-015-0028-1
Scott BL, Evans RW, Jankovic J. Treatment of palatal myoclonus with sumatriptan. Mov Disord. 1996;11:748–751.
Nasr A, Brown N. Palatal myoclonus responding to lamotrigine. Seizure. 2002;11:136–137.
Obeso JA. Therapy of myoclonus. Clin Neurosci. 1995;3(4):253–7.
Krause E, Leunig A, Klopstock T, Gürkov R. Treatment of essential palatal myoclonus in a 10-year-old girl with botulinum neurotoxin. Otol Neurotol. 2006;27(5):672–675.
Srirompotong S, Tiamkao S, Jitpimolmard S. Botulinum toxin injection for objective tinnitus from palatal myoclonus: a case report. J Med Assoc Thai. 2002;85:392–395.
Penney SE, Bruce IA, Saeed SR. Botulinum toxin is effective and safe for palatal tremor: a report of five cases and a review of the literature. J Neurol. 2006;253:857–860.
Sinclair CF, Gurey LE, Blitzer A. Palatal myoclonus: algorithm for management with botulinum toxin based on clinical disease characteristics. Laryngoscope. 2014;124(5):1164–1169.
Ensink RJ, Vingerhoets HM, Schmidt CW, Cremers CW. Treatment for severe palatoclonus by occlusion of the Eustachian tube. Otol Neurotol. 2003;24(5):714–716.
Baik JS, Lyoo CH, Lee JH, Lee MS. Drug-induced and psychogenic resting suprahyoid neck and tongue tremors. Movement Disord 2008; 23:746–748.
Williams DR. Psychogenic palatal tremor. Mov Disord.2004; 19: 333–5.
Yokota T, Hirashima F, Ito Y, Tanabe H, Furukawa T, Tsukagoshi H. Idiopathic palatal myoclonus. Acta Neurol Scand. 1990; 81: 239–42.
Keswani SC, Kossoff EH, Krauss GL, Hagerty C. Amelioration of spinal myoclonus with levetiracetam. J Neurol Neurosurg Psychiatry. 2002;73(4):457–458.
Estraneo A, Saltalamacchia AM, Loreto V. Spinal myoclonus following herpes zoster radiculitis. Neurology. 2007;68:E4.
Campos CR, Limongi JC, Machado FC, Brotto MW. A case of primary spinal myoclonus: clinical presentation and possible mechanisms involved. Arq Neuropsiquiatr. 2003;61(1):112-114. doi:https://doi.org/10.1590/s0004-282x2003000100022
Polo KB, Jabbari B. Effectiveness of botulinum toxin type A against painful limb myoclonus of spinal cord origin. Mov Disord. 1994;9(2):233-235. doi:https://doi.org/10.1002/mds.870090221
Lagueny A, Tison F, Burbaud P, Le Masson G, Kien P. Stimulus-sensitive spinal segmental myoclonus improved with injections of botulinum toxin type A. Mov Disord. 1999;14(1):182-185. doi:https://doi.org/10.1002/1531-8257(199901)14:1<182::aid-mds1040>3.0.co;2-8
Yoshimura DM, Aminoff MJ, Tami TA, Scott AB. Treatment of hemifacial spasm with botulinum toxin. Muscle Nerve. 1992;15(9):1045–1049.
Alexander GE, Moses 3rd H. Carbamazepine for hemifacial spasm. Neurology. 1982;32(3):286–7.
Daniele O, Caravaglios G, Marchini C, Mucchiut L, Capus P, Natalè E. Gabapentin in the treatment of hemifacial spasm. Acta Neurol Scand. 2001;104(2):110–112.
Kaye AH, Adams CB. Hemifacial spasm: a long term follow-up of patients treated by posterior fossa surgery and facial nerve wrap**. J Neurol Neurosurg Psychiatry. 1981;44(12):1100–3.
Janetta PJ. Vascular compression of the facial nerve at the brainstem in hemifacial spasm: treatment by microsurgical decompression. In: Morley TP, ed. Current Controversies in Neurosurgery. Philadelphia: WB Saunders, 1976.
Kamble NL, Pal PK. Electrophysiological evaluation of psychogenic movement disorders. Parkinsonism Relat Disord. 2016;22 Suppl 1:S153-S158. doi:https://doi.org/10.1016/j.parkreldis.2015.09.016a
Beudel M, Zutt R, Meppelink AM, et al. Improving neurophysiological biomarkers for functional myoclonic movements. Parkinsonism Relat Disord. 2018;51:3-8. doi:https://doi.org/10.1016/j.parkreldis.2018.03.029
Czarnecki K, Hallett M. Functional (psychogenic) movement disorders. Curr Opin Neurol. 2012;25(4):507–512.
Dreissen YEM, Cath DC, Tijssen MAJ. Functional jerks, tics, and paroxysmal movement disorders. Handb Clin Neurol. 2016;139:247–258.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 506 kb)
Rights and permissions
About this article

Cite this article
Pena, A.B., Caviness, J.N. Physiology-Based Treatment of Myoclonus. Neurotherapeutics 17, 1665–1680 (2020). https://doi.org/10.1007/s13311-020-00922-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-020-00922-6