
Abstract
Purpose
Deficiency of adenosine deaminase 2 (DADA2) is an autosomal recessive disorder that manifests with fever, early-onset vasculitis, strokes, and hematologic dysfunction. This study aimed to identify disease-causing variants by conventional Sanger and whole exome sequencing in two families suspected to have DADA2 and non-confirmatory genotypes. ADA2 enzymatic assay confirmed the clinical diagnosis of DADA2. Molecular diagnosis was important to accurately identify other family members at risk.
Methods
We used a variety of sequencing technologies, ADA2 enzymatic testing, and molecular methods including qRT-PCR and MLPA.
Results
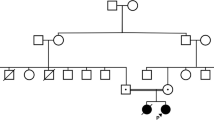
Exome sequencing identified heterozygosity for the known pathogenic variant ADA2: c.1358A>G, p.Tyr453Cys in a 14-year-old female with a history of ischemic strokes, livedo, and vasculitis. No second pathogenic variant could be identified. ADA2 enzymatic testing in combination with quantitative RT-PCR suggested a loss-of-function allele. Subsequent genome sequencing identified a canonical splice site variant, c.-47+2T>C, within the 5′UTR of ADA2. Two of her unaffected siblings were found to carry the same two pathogenic variants. A homozygous 800-bp duplication comprising exon 7 of ADA2 was identified in a 5-year-old female with features consistent with Diamond-Blackfan anemia (DBA). The duplication was missed by Sanger sequencing of ADA2, chromosomal microarray, and exome sequencing but was detected by MLPA in combination with long-read PCR sequencing. The exon 7 duplication was also identified in her non-symptomatic father and younger sister.
Conclusions
ADA2 pathogenic variants may not be detected by conventional sequencing and genetic testing and may require the incorporation of additional diagnostic methods. A definitive molecular diagnosis is crucial for all family members to make informed treatment decisions.



Similar content being viewed by others


References
Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370(10):911–20.
Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med. 2014;370(10):921–31.
Hashem H, Egler R, Dalal J. Refractory pure red cell aplasia manifesting as deficiency of adenosine deaminase 2. J Pediatr Hematol Oncol. 2017 Jul;39(5):e293–6.
Hashem H, Kumar AR, Muller I, Babor F, Bredius R, Dalal J, et al. Hematopoietic stem cell transplantation rescues the hematological, immunological, and vascular phenotype in DADA2. Blood. 2017;130(24):2682–8.
Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. 2016;177:316–20.
Schepp J, Bulashevska A, Mannhardt-Laakmann W, Cao H, Yang F, Seidl M, et al. Deficiency of adenosine deaminase 2 causes antibody deficiency. J Clin Immunol. 2016;36(3):179–86.
Schepp J, Proietti M, Frede N, Buchta M, Hübscher K, Restrepo JR, et al. Screening of 181 patients with antibody deficiency for deficiency of adenosine deaminase 2 sheds new light on the disease in adulthood. Arthritis Rheum. 2017;69(8):1689–700.
Özen S, Batu ED, Taşkıran EZ, Özkara HA, Ünal Ş, Güleray N, et al. A monogenic disease with a variety of phenotypes: deficiency of adenosine deaminase 2. J Rheumatol. 2020;47(1):117–25.
Van Nieuwenhove E, Humblet-Baron S, Van Eyck L, De Somer L, Dooley J, Tousseyn T, et al. ADA2 deficiency mimicking idiopathic multicentric Castleman disease. Pediatrics. 2018;142(3).
Nanthapisal S, Murphy C, Omoyinmi E, Hong Y, Standing A, Berg S, et al. Deficiency of adenosine deaminase type 2: a description of phenotype and genotype in fifteen cases. Arthritis Rheum. 2016;68(9):2314–22.
Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (Oxford). 2016;55(5):902–10.
Springer JM, Gierer SA, Jiang H, Kleiner D, Deuitch N, Ombrello AK, et al. Deficiency of adenosine deaminase 2 in adult siblings: many years of a misdiagnosed disease with severe consequences. Front Immunol. 2018;9(1361).
Batu ED, Karadag O, Taskiran EZ, Kalyoncu U, Aksentijevich I, Alikasifoglu M, et al. A case series of adenosine deaminase 2-deficient patients emphasizing treatment and genotype-phenotype correlations. J Rheumatol. 2015;42(8):1532–4.
Claassen D, Boals M, Bowling KM, Cooper GM, Cox J, Hershfield M, et al. Complexities of genetic diagnosis illustrated by an atypical case of congenital hypoplastic anemia. Cold Spring Harb Mol Case Stud. 2018;4(6):a003384.
Caorsi R, Penco F, Grossi A, Insalaco A, Omenetti A, Alessio M, et al. ADA2 deficiency (DADA2) as an unrecognised cause of early onset polyarteritis nodosa and stroke: a multicentre national study. Ann Rheum Dis. 2017;76(10):1648–56.
Aksentijevich I, Moura NS, Barron K. Adenosine deaminase 2 deficiency. GeneReviews®[Internet]: University of Washington, Seattle; 2019.
Gibson KM, Morishita KA, Dancey P, Moorehead P, Drogemoller B, Han X, et al. Identification of novel adenosine deaminase 2 gene variants and varied clinical phenotype in pediatric vasculitis. Arthritis & rheumatology (Hoboken, NJ). 2019.
Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J Allergy Clin Immunol. 2020;13.
Hashem H, Vatsayan A, Gupta A, Nagle K, Hershfield M, Dalal J. Successful reduced intensity hematopoietic cell transplant in a patient with deficiency of adenosine deaminase 2. Bone Marrow Transplant. 2017;52(11):1575–6.
Van Eyck L Jr, Hershfield MS, Pombal D, Kelly SJ, Ganson NJ, Moens L, et al. Hematopoietic stem cell transplantation rescues the immunologic phenotype and prevents vasculopathy in patients with adenosine deaminase 2 deficiency. J Allergy Clin Immunol. 2015;135(1):283–287.e285.
Ombrello A, Stone D, Hoffmann P, Jones A, Barham B, Barron K, et al. The deficiency of adenosine deaminase type 2-results of therapeutic intervention. Pediatr Rheumatol Online J. 2015;13(Suppl 1):O40.
Ombrello AK, Qin J, Hoffmann PM, Kumar P, Stone D, Jones A, et al. Treatment strategies for deficiency of adenosine deaminase 2. N Engl J Med. 2019;380(16):1582–4.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101.
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461–8.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. 2019:531210.
Chong-Neto HJ, Segundo GRS, Bandeira M, Chong-Silva DC, Rosário CS, Riedi CA, et al. Homozygous splice ADA2 gene mutation causing ADA-2 deficiency. J Clin Immunol. 2019;39(8):842–5.
Trotta L, Martelius T, Siitonen T, Hautala T, Hamalainen S, Juntti H, et al. ADA2 deficiency: clonal lymphoproliferation in a subset of patients. J Allergy Clin Immunol. 2018;141(4):1534–1537.e1538.
Alsultan A, Basher E, Alqanatish J, Mohammed R, Alfadhel M. Deficiency of ADA2 mimicking autoimmune lymphoproliferative syndrome in the absence of livedo reticularis and vasculitis. Pediatr Blood Cancer. 2018;65(4).
Clark MJ, Chen R, Lam HYK, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29(10):908–14.
Shigemizu D, Momozawa Y, Abe T, Morizono T, Boroevich KA, Takata S, et al. Performance comparison of four commercial human whole-exome capture platforms. Sci Rep. 2015;5(1):12742.
Chilamakuri CSR, Lorenz S, Madoui M-A, Vodák D, Sun J, Hovig E, et al. Performance comparison of four exome capture systems for deep sequencing. BMC Genomics. 2014;15(1):449.
Asan XY, Jiang H, Tyler-Smith C, Xue Y, Jiang T, et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12(9):R95.
Grossi A, Cusano R, Rusmini M, Penco F, Schena F, Podda RA, et al. ADA2 deficiency due to a novel structural variation in 22q11.1. Clin Genet. 2019;95(6):732–3.
Song X, Beck CR, Du R, Campbell IM, Coban-Akdemir Z, Gu S, et al. Predicting human genes susceptible to genomic instability associated with Alu/Alu-mediated rearrangements. Genome Res. 2018;28(8):1228–42.
Weckselblatt B, Rudd MK. Human structural variation: mechanisms of chromosome rearrangements. Trends Genet. 2015;31(10):587–99.
Conceição Pereira M, Loureiro JL, Pinto-Basto J, Brandão E, Margarida Lopes A, Neves G, et al. Alu elements mediate large SPG11 gene rearrangements: further spatacsin mutations. Genetics in Medicine. 2012;14(1):143–51.
Gu W, Zhang F, Lupski JR. Mechanisms for human genomic rearrangements. Pathogenetics. 2008;1(1):4–4.
Liebowitz J, Hellmann DB, Schnappauf O, et al. J Rheumatol. 2019; jrheum.180820.
Yao R, Zhang C, Yu T, Li N, Hu X, Wang X, et al. Evaluation of three read-depth based CNV detection tools using whole-exome sequencing data. Mol Cytogenet. 2017;10:30.
Chiang T, Liu X, Wu T-J, Hu J, Sedlazeck FJ, White S, et al. Atlas-CNV: a validated approach to call single-exon CNVs in the eMERGESeq gene panel. Genet Med. 2019;21(9):2135–44.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011 Jun;21(6):974–84.
Fromer M, Moran JL, Chambert K, Banks E, Bergen SE, Ruderfer DM, et al. Discovery and statistical genoty** of copy-number variation from whole-exome sequencing depth. Am J Hum Genet. 2012;91(4):597–607.
Erzurumluoglu AM, Shihab HA, Rodriguez S, Gaunt TR, Day INM. Importance of genetic studies in consanguineous populations for the characterization of novel human gene functions. Ann Hum Genet. 2016;80(3):187–96.
Bittles AH, Black ML. Consanguinity, human evolution, and complex diseases. Proc Natl Acad Sci. 2010;107(suppl 1):1779–86.
Acknowledgments
We would like to thank the patients, the families, and the healthy controls for their enthusiastic support during this study. We also thank Adelani Adeleye, Camille Alba, Dagmar Bacikova, Daniel N. Hupalo, Elisa McGrath Martinez, Anthony R. Soltis, Gauthaman Sukumar, and **jun Zhang from the American Genome Center (Uniformed Services University of the Health Sciences, Bethesda, MD, USA) and Harvey B. Pollard and Matthew D. Wilkerson from the Department of Anatomy, Physiology & Genetics (Uniformed Services University of the Health Sciences, Bethesda, MD, USA).
Funding
This research was financially supported by the Intramural Research Programs of the NHGRI and the NIH Clinical Center. The study was additionally supported by a NIDCR, NIH intramural research grant (1ZIADE000695) to JAC and a DDIR Innovation Award to JAC and DGM. Genome sequencing was supported by the Department of Defense (award W81XWH-09-2-0128). This work utilized the computational resources of the NIH HPC Biowulf cluster. OS is supported by the German Research Foundation.
Author information
Authors and Affiliations
Contributions
OS conceived the study, performed experiments, analyzed data, and drafted the manuscript. QZ, NSM, ND, and MH performed experiments, analyzed the data. DGW and JAC performed RNA sequencing and analysis. CA, HTB, DBB, PDW, NS, KB, DLB, PH, and DLK recruited patients and revised the manuscript. EW, AKO, and IA provided crucial conceptual inputs and helped in writing the manuscript. TAGC, CLD, and NISC performed Next Generation Sequencing and data analyses. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
The study was approved by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK)/National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) Institutional Review Board.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic Supplementary Material
ESM 1
(PDF 272 kb)
Rights and permissions
About this article

Cite this article
Schnappauf, O., Zhou, Q., Moura, N.S. et al. Deficiency of Adenosine Deaminase 2 (DADA2): Hidden Variants, Reduced Penetrance, and Unusual Inheritance. J Clin Immunol 40, 917–926 (2020). https://doi.org/10.1007/s10875-020-00817-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-020-00817-3