
Abstract
Purpose
Severe Combined Immune Deficiency (SCID) is universally fatal unless treated with hematopoietic stem cell transplantation (HSCT). Following the identification of disseminated Bacille Calmette-Guérin (BCG) infections in Canadian First Nations, Métis and Inuit (FNMI) children with unrecognized primary immune deficiencies, a national surveillance study was initiated in order to determine the incidence, diagnosis, treatment and outcome of children with SCID in Canada.
Methods
Canadian pediatricians were asked to complete a monthly reporting form if they had seen a suspected SCID case, from 2004 to 2010, through the Canadian Paediatric Surveillance Program (CPSP). If the case met CPSP SCID criteria, more detailed data, including demographics and clinical information about investigations, treatment and outcome was collected.
Results
A total of 40 cases of SCID were confirmed for an estimated incidence of SCID in non-FNMI Canadian children of 1.4 per 100,000 live births (95 % CI 1 to 1.9/100,000). The proportion of SCID cases that were FNMI (17.5 %) was almost three times higher than was expected on the basis of proportion of the pediatric population estimated to be FNMI (6.3 %) resulting in an estimated incidence of 4.4 per 100,000 live births (95 % CI 2.1 to 9.2/100,000) in FNMI Canadian children. The mean age at diagnosis for all SCID cases was 4.2 months (range 1–583 days). There were 12 deaths (30 %; 95 % CI 18–46 %); seven died of confirmed or suspected infections before they could receive an HSCT.
Conclusions
The frequency of SCID cases in FNMI children is higher than in the general Canadian pediatric population. The high mortality rate, due primarily to infection, suggests that early diagnosis by newborn screening followed by HSCT could significantly benefit children with SCID.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Severe combined immunodeficiency (SCID) is a rare primary immune deficiency caused by genetic mutations resulting in severe deficiencies in both cellular and humoral immunity. SCID patients are susceptible to serious infections and will typically die before 1 year of age if immune function is not restored through allogeneic hematopoietic stem cell transplantation (HSCT), enzyme replacement or gene therapy [1]. SCID is a pediatric emergency and early diagnosis is essential in order to initiate life-saving therapies and avoid severe infections and organ toxicity.
In 2003, public health authorities became aware of a number of cases of vaccine-associated disseminated BCG mycobacterial infection reported through the IMPACT (Immunization Program Monitoring Active) surveillance network [2]. Eight of the nine cases identified were in First Nations, Métis and Inuit (FNMI) children and seven of those children had an underlying genetic immune deficiency (5 SCID, 1 interferon gamma receptor deficiency and 1 uncharacterized) [3]. Disseminated BCG disease is a rare but devastating complication of BCG vaccination in severely immune compromised infants. This lead to a re-evaluation of routine BCG vaccination practices in First Nations and Inuit communities. At the time, the frequency of SCID in Canada was unknown and Health Canada’s First Nations and Inuit Health Branch (FNIHB) initiated a national surveillance study in collaboration with the Canadian Pediatric Society and pediatric academic centers across Canada. Acquiring this information was crucial to public health policy decision-making regarding the continued use of BCG vaccination in First Nations communities and the need for SCID newborn screening in Canada.
This study used the framework of the Canadian Paediatric Surveillance Program (CPSP), in which data are gathered from over 2,500 Canadian paediatricians and paediatric subspecialists each month.
The objectives of this study were to determine: 1) the incidence and type of SCID in the Canadian FNMI and general populations; 2) the incidence of disseminated BCG disease in SCID patients; 3) the average time delay to diagnosis; 4) known risk factors and initial clinical presentations; 5) whether patients received a HSCT or gene therapy; and 6) the causes of any deaths.
Methods
Potential cases were identified through the CPSP from April 1, 2004 to March 31, 2010. A collaboration of the Canadian Paediatric Society and the Public Health Agency of Canada, this program maintains monthly contact with more than 2,500 paediatricians and paediatric subspecialists in a two-tiered reporting system [4]. The physician indicates that they have seen the disease in the last month or submits a “nil report”. In follow-up, the CPSP sends a detailed data collection form for reporting physicians to complete. The detailed reporting form for a SCID case is available online at the Canadian Paediatric Society website [5]. In almost all cases, the information provided in the reporting form was incomplete due to absence or lack of reporting. When the data were insufficient to make a definitive diagnosis the study investigator requested additional information, if available, from the CPSP who mediated requests to reporting physicians. However because of incomplete reporting of all the demographic and clinical variables, results do not equal the total number of reported cases in all analyses.
Ethical approval for this national study was obtained from the Health Canada Research Ethics Board and Research Ethics Board at IWK Health Centre in Halifax, Canada. This study was preformed in accordance with the 1964 Declaration of Helsinki and its later amendments.
Inclusion and Exclusion Criteria
The CPSP case definition for SCID was any child less than 2 years of age with the clinical features of SCID (including chronic diarrhea, recurrent pneumonia, failure to thrive, persistent thrush, and opportunistic infections) and at least one of the following: 1) an absolute lymphocyte count of less than 3000/mm3 or less than 20 % CD3+ T cells, 2) familial history of primary immunodeficiency. Infants with HIV infection or cystic fibrosis were excluded. This case definition for SCID was less specific than the diagnostic criteria for SCID put forth in 1999 by PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) and updated in 2005, which requires additional criteria such as engraftment of trans-placental-acquired maternal T cells, a definitive genetic mutation or decreased proliferative responses to mitogens [6]. This was done in order to capture as many cases as possible. Two central study investigators (JR and AJ) reviewed each CPSP case report to determine if it was a definitive or probable case of SCID based on PAGID/ESID criteria.
Results
The Incidence and Types of SCID in Canadian Children
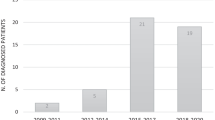
The average annual national reporting rate during the study period for the initial questionnaire was 80.4 % ± SD 1.4 % and the average annual number of participants was 2560 ± SD 43. The completion rate for the detailed SCID questionnaire was 90 %. The CPSP received 102 suspected SCID case reports. Of these 62 were either duplicates or excluded according to the CPSP case definition. A total of 40 cases met the CPSP case definition for SCID during the study period (2004 to 2010).
Of the 40 cases meeting the CPSP case definition for SCID, 32 (80 %) fulfilled ESID criteria for definitive or probable SCID or X-linked SCID. Evidence of deficient ADA catalytic activity and/or causative genetic mutations were identified in 20 of the 32 cases: adenosine deaminase (ADA) deficiency (n = 10), gamma chain deficiency (n = 4) and RAG1 deficiency (n = 2). There were also 2 cases each of ZAP70 and MHC Class II mutations. ZAP70 and MHC class II deficiencies are not considered classical SCID [7] but in all four cases, the patients presented with clinical findings of typical SCID. The remaining 12 of 32 cases were assigned per ESID diagnostic criteria as probable SCID (n = 9), definitive X-linked SCID (n = 1), probable X-linked SCID (n = 1) and possible X-linked SCID (n = 1). Of the remaining 8 of 40 that did not fulfill ESID criteria but fulfilled the CPSP case definition, three were transplanted for presumed SCID, two were waiting for transplant and two had passed away.
We included all 40 cases in our calculation of incidence. Of the 40 confirmed CPSP cases, seven were FNMI children, a disproportionate number (17.5 %; 95 % CI 10.7–24.3 %) given that FNMI children represent 6.3 % of the total Canadian pediatric population based on the Statistics Canada 2006 census of the 0–14 age group [8]. This larger proportion is likely to introduce a bias in our incidence calculation and we therefore calculated the incidence in FNMI and non-FNMI Canadian children separately.
During the study period (2004 to 2010) 2,537,691 live births were recorded in Canada [9]; 39 of 40 patients were born in Canada. If we predict that 6.3 % of those live births were FNMI children, the estimated incidence of SCID in FNMI and non-FNMI Canadian children is 4.4 per 100,000 live births (95 % CI 2.1 to 9.2/100,000) and 1.4 per 100,000 live births (95 % CI 1 to 1.9/100,000) respectively.
For the entire cohort, the mean age at diagnosis was 125 days (mean 4.2 months; range 1–583 days) based on 32 of 40 cases reporting date of diagnosis or time of onset of clinical manifestations of SCID.
Evaluation of Absolute Lymphocyte Count and Risk Factors in SCID Patients
A commonly applied test for diagnosis of SCID is the absolute lymphocyte count (ALC) with an ALC of <2500/mm3 as potentially pathogenic in early infancy [10]. At risk populations can also be identified by evaluation of risk factors including a family history of SCID and/or infant deaths and consanguinity. We evaluated whether the ALC met this criteria in the Canadian SCID population. The mean ALC at diagnosis was 2579/mm3 (median: 1130/mm3; range: 50–14,040/mm3), (32 of 40 confirmed cases). Important clinical clues such as family history of SCID, infant death and/or consanguinity were present in 17 of 32 cases (53 %) reporting on family history. Specifically in the FNMI cohort of patients, 3 of 7 had a positive family history and/or consanguinity (43 %). Among 12 cases with no relevant family history and a reported ALC, eight had an ALC < 2500/mm3. Screening with both the ALC and risk factors properly identified 25 of the 32 confirmed cases (78 %).
Clinical Symptoms at Presentation in Canadian Children with SCID
The three most common presenting clinical manifestations were: interstitial pneumonia (48 %), failure to thrive (43 %), and a persistent bronchiolitic-like illness (40 %) (Table I). In 36 of the 40 SCID cases, one or more infections had been diagnosed at the time of reporting, including bacterial, viral and fungal pathogens (Table II). No patients reported in this series were given BCG and the study did not capture any cases of disseminated BCG infection.
Therapeutic Interventions in Treated Canadian SCID Patients
Of the 18 patients with recorded information, 15 patients received a hematopoietic stem cell transplantation (HSCT), two received enzyme replacement therapy for ADA SCID and one was referred for gene therapy. Of the 15 patients who were transplanted, 6 had no family history (40 %). There was no statistical difference in average days to diagnosis between BMT patients with a family history and those without, 123 versus 110 days respectively. Those without a family history were identified due to clinical features of SCID. Among the 15 transplant patients, there was a fatal outcome in three (20 %) due to GVHD (n = 2) and respiratory failure (n = 1). The donor sources for the transplant were matched unrelated (n = 7), matched sibling (n = 4), haploidentical related (n = 2) and unknown (n = 2).
Mortality in our Canadian SCID Cohort
There was a mortality rate of 30 % (12/40 cases) in the entire SCID population. Detailed information on each death is provided in Table III. The mean age at diagnosis based on 11/12 cases was 96 days (3.2 months). Four patients died before they could be referred for HSCT and another three patients died while waiting for a HSCT. Infections were the confirmed cause of death in four cases (disseminated CMV (n = 3), H1N1 Influenza A (n = 1)) and highly suspected in another 3 cases (ARDS (n = 1), respiratory failure (n = 1) and encephalitis (n = 1)).
Discussion
This is the first national study in Canada to estimate the incidence of SCID. Based on the study results, an observed incidence rate of 1.4 in 100,000 live births is reported in the Canadian non-FNMI population, which is consistent with previous studies from the US and France [11, 12].
The disproportionate higher estimated incidence of FNMI SCID cases, 4.4 in 100,000 live births, suggests that there is a skewed distribution of SCID in Canada. This may be due to founder effects caused by ancestral mutations in certain Aboriginal populations similar to the high frequency of T-B- SCID among Athabascan-speaking Native Americans [13]. In our FNMI cohort, we did not find a higher incidence of positive family history and/or consanguinity compared to the larger group, although the numbers are small. In order to calculate the incidence among FNMI children we estimated the number of FNMI births based on the FNMI percentage of the Canadian pediatric population using census data that reported the number of people stating Aboriginal identity among different age groups, as there are no complete statistics on birthrates among FNMI peoples of Canada.
We could not comment on the incidence of disseminated BCG infection, as none of the patients reported in this study were administered BCG vaccine. This could be due to the fact that the use of BCG vaccine, even in First Nations communities, has gradually been discontinued across Canada in the last 10 years and replaced with enhanced Tuberculosis screening and control services. For example, the routine use of BCG was discontinued in First Nations communities in British Columbia and Quebec in 2003 and 2005, respectively [14].
Hematopoietic stem cell transplantation (HSCT) performed early before significant infections have developed has been shown to result in the optimal survival for a majority of SCID infants. In one series of SCID patients, those receiving a transplant before 106 days (3.5 months of age), have a survival rate of 95 % compared to 76 % for those who received transplants when they were 3.5 months or older [15]. The average days at diagnosis in the current report for the confirmed cases was 125 days (4.2 months of age), despite positive risk factors and characteristic clinical signs in many instances. The average days at diagnosis for those patients that died was even shorter at 96 days. Seven patients died of infections before they could receive an HSCT. Thus, earlier diagnosis followed by hematopoietic stem cell transplantation could potentially decrease the mortality in our population.
It is well established that neonatal diagnosis of SCID leads to significantly improved survival outcomes [16]. Diagnosis at birth could prevent the almost certain life-threatening infections and end-organ damage, thus improving transplant outcome. In our report, survival after transplant was 80 % (12/15 cases). However, key risk factors such as family history of SCID and infant death and consanguinity were only present in half of the reported cases. This has been the impetus for establishing neonatal screening programs for SCID. The first public health program to establish screening was in the state of Wisconsin, USA in 2008. Currently, half of all births in the United States are being screened for SCID as part of pilot or established state newborn screening programs. The standard approach for SCID screening is testing for T-cell receptor excision circles (TRECs), a DNA biomarker of normal T-cell development. They can be measured in DNA isolated from the dried bloodspots already collected for newborn screening [17]. TREC screening will not detect primary immunodeficiencies in the differential diagnosis of SCID such as ZAP70 and MHC class II and certain cases of ADA deficiency, but it does have the potential to detect other SCID-related and T lymphopenic disorders not captured in this study [18]. Universal screening for SCID has been predicted to be a cost effective means to improve quality and duration of life for children with SCID [19], but currently there is only one provincial neonatal screening program for SCID in Canada in Ontario. We believe that SCID fulfills requirements for addition to newborn screening panels in Canada. These criteria include: 1) SCID is fatal, often in the first year of life; 2) Newborns with SCID usually appear healthy at birth; 3) our estimated incidences support screening; 4) diagnostic testing is well-established; 5) curative treatment exists and earlier treatment leads to better outcomes; and 6) lastly there is a cost-effective and accurate screening tool.
There are several limitations to this national surveillance study. Investigators had no control over how the original data were collected and we acknowledge that some SCID cases may have been missed because of the exclusion criteria and general rate of participation in the CPS Surveillance Program. We do not know the exact fraction of the Canadian infant population that was captured by this surveillance study or the percentage of Canadian pediatricians and pediatric subspecialists represented in this study but we are encouraged by the national reporting rate of 80 % and high number of duplicate reports. The inclusion criteria we used was not as vigorous as those used by PAGID or ESID and in particular the use of an ALC < 3000/mm3 may have failed to capture patients with maternal T cell engraftment. This could also explain why our fraction of ADA deficiency cases was higher then expected. In contrast to other forms of SCID, maternal lymphoid engraftment is not observed with ADA deficiency [20]. Another possibility is the existence of founder mutations for ADA deficiency and ZAP70 SCID in Canadian old colony Mennonite populations. Unfortunately, this study did not capture any information about ethnicity beyond FNMI status. The use of a fixed ALC can also be problematic as the normal lymphocyte range is very age dependent. We believe that the current diagnostic criteria set forth by the Primary Immune Deficiency Treatment Consortium (PIDTC) as part of their prospective SCID study 6901 is the most rigorous description to date and should be employed in future studies [21]. The reporting form often had missing data in certain categories. Because of the many variables involved in the analysis, each with data missing for a number of cases, this can lead to biased results. Information reporting tends to favor interesting and unusual results rather then normal ones. This study further highlights the need for Canadian centers to register their primary immunodeficiency patients in registries such as the North American United States Immunodeficiency Network (USIDNET) and participate in multi-center SCID studies as part of the PIDTC in order to acquire more comprehensive data.
Conclusions
This national cohort study provides the first report on the incidence and outcome of SCID patients in Canada. It provides evidence that the incidence is comparable to other national studies, but there is a higher percentage in the First Nations, Metis and Inuit pediatric population. It is clear based on recorded deaths, the majority due to infectious complications, that early identification of SCID is critical to improved survival. We argue that the results of this study provide compelling evidence that the case for universal SCID newborn screening in Canada needs to be a high priority for public health decision markers in Canada.
References
Sponzilli I, Notarangelo LD. Severe combined immunodeficiency (SCID): from molecular basis to clinical management. Acta Biomed. 2011;82(1):5–13.
Deeks SL, Clark M, Scheifele DW, et al. Serious adverse events associated with bacille Calmette-Guérin vaccine in Canada. Pediatr Infect Dis J. 2005;24(6):538–41.
Dawar M, Clark M, Deeks SL, et al. A fresh look at an old vaccine: does BCG have a role in 21st century Canada? Int J Circumpolar Health. 2004;63 Suppl 2:230–6.
Ugnat AM, Grenier D, Thibodeau ML, et al. The Canadian paediatric surveillance program: celebrating 15 years of successful paediatric surveillance. Paediatr Child Health. 2011;16(4):203–5.
http://www.cpsp.cps.ca/uploads/studies/severe-combined-immunodeficiency-questionnaire.pdf. Accessed 10 July 2013.
Conley M, Notarangelo L, Etzioni A, Representing PAGID, Pan-American Group for Immunodeficiency, and ESID, European Society for Immunodeficiencies. Diagnostic criteria for primary immunodeficiencies. Clin Immunol. 1999;93(3):190–7.
Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification form the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2:54. doi:10.3389/fimmu.2011.00054. Epub 2011 Nov 8.
Statistics Canada. Table 102-4509 - Live births, by birth weight and sex, Canada, provinces and territories, annual, CANSIM (database)
Statistics Canada. Aboriginal identity population by age groups, median age and sex, 2006 counts for both sexes, for Canada, provinces and territories - 20 % sample data (table). 2006. http://www12.statcan.ca/census-recensement/2006/dp-pd/hlt/97-558/pages/page.cfm?Lang=E&Geo=PR&Code=01&Table=1&Data=Count&Sex=1&Age=2&StartRec=1&EndRec=13&Sort=2&Display=All&CSDFilter=250 (accessed September 24, 2013).
Adeli MM, Buckley RH. Why newborn screening for severe combined immunodeficiency is essential: a case report. Pediatrics. 2010;126(2):e465–9.
Chan K, Puck JM. Development of population-based newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2005;115(2):391–8.
Stephan JL, Vlekova V, Le Deist F, et al. Severe combined immunodeficiency: a retrospective single-center study of clinical presentation and outcome in 117 patients. J Pediatr. 1993;123(4):564–72.
Li L, Moshous D, Zhou Y, et al. A founder mutation in Artemis, an SNM1-like protein, causes SCID in Athabascan-speaking Native Americans. J Immunol. 2002;168(12):6323–9.
Phac-aspc.gc.ca. BCG Vaccine Usage in Canada - Current and Historical - Tuberculosis Prevention and Control - PHAC. 2012. [online] Available at: http://www.phac-aspc.gc.ca/tbpc-latb/bcgvac_1206-eng.php [Accessed: 7 Jan 2013].
Buckley RH, Schiff SE, Schiff RI, et al. Hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 1999;340(7):508–16.
Brown L, Xu-Bayford J, Allwood Z, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117(11):3243–6.
Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. J Allergy Clin Immunol. 2012;129(3):607–16.
Kwan A, Church JA, Cowan MJ, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132(1):140–50.
Chan K, Davis J, Pai SY, et al. A Markov model to analyze cost-effectiveness of screening for severe combined immunodeficiency (SCID). Mol Genet Metab. 2011;104(3):383–9.
Buckley RH, Schiff SE, Schiff RI, et al. Human severe combined immunodeficiency: genetic, phenotypic, and functional diversity in one hundred eight infants. J Pediatr. 1997;130(3):378–87.
Dvorak CC, Cowan MJ, Logan BR, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013 Jul 2. [Epub ahead of print].
Acknowledgments
We would like to thank Ms. Ruth Milner for her statistical support and Dr. Louise Pelletier, Dr. Maura Ricketts, Dr. Marcus Lem and Dr. Ezzat Farzad for their help in setting up the study. Funding for this project was obtained from the Office of Community Medicine at FNIHB, Health Canada.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rozmus, J., Junker, A., Thibodeau, M.L. et al. Severe Combined Immunodeficiency (SCID) in Canadian Children: A National Surveillance Study. J Clin Immunol 33, 1310–1316 (2013). https://doi.org/10.1007/s10875-013-9952-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10875-013-9952-8