
Abstract
Bile acids are a group of chemically different steroids generated at the host/microbial interface. Indeed, while primary bile acids are the end-product of cholesterol breakdown in the host liver, secondary bile acids are the products of microbial metabolism. Primary and secondary bile acids along with their oxo derivatives have been identified as signaling molecules acting on a family of cell membrane and nuclear receptors collectively known as “bile acid-activated receptors.” Members of this group of receptors are highly expressed throughout the gastrointestinal tract and mediate the bilateral communications of the intestinal microbiota with the host immune system. The expression and function of bile acid-activated receptors FXR, GPBAR1, PXR, VDR, and RORγt are highly dependent on the structure of the intestinal microbiota and negatively regulated by intestinal inflammation. Studies from gene ablated mice have demonstrated that FXR and GPBAR1 are essential to maintain a tolerogenic phenotype in the intestine, and their ablation promotes the polarization of intestinal T cells and macrophages toward a pro-inflammatory phenotype. RORγt inhibition by oxo-bile acids is essential to constrain Th17 polarization of intestinal lymphocytes. Gene-wide association studies and functional characterizations suggest a potential role for impaired bile acid signaling in development inflammatory bowel diseases (IBD). In this review, we will focus on how bile acids and their receptors mediate communications of intestinal microbiota with the intestinal immune system, describing dynamic changes of bile acid metabolism in IBD and the potential therapeutic application of targeting bile acid signaling in these disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The inflammatory bowel diseases (IBD), encompassing Crohn’s disease (CD), ulcerative colitis (UC), are chronic relapsing disorders caused by a dysregulated intestinal immune response to luminal antigens in genetically predisposed subjects that have emerged as a public health challenge worldwide. Traditionally regarded as a disease of westernized nations, the prevalence of IBD is rising rapidly worldwide among ethnicities and nationalities in whom IBD were previously uncommon [1]. While changes in the composition of the intestinal microbiota are recognized as the main drivers of dysregulated intestinal immunity seen in IBD, the chemical mediators that support this altered communication are still incompletely defined. Bile acids are steroidal molecules generated at the interface of the host and the intestinal microbiota [2]. Together, they represent one of the largest family of biologically active mediators found in mammalians acting on family of G-protein and nuclear receptors (Fig. 1 and Table 1). In this article, we will review the role of these steroids as signaling molecules placed at the interface between the intestinal microbiota and host immune system and how dysregulation of bile acid signaling might have mechanistic relevance in the development of IBD.

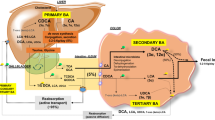
Hepatic bile acid metabolism. a Bile acids are synthesized in the liver from cholesterol by two metabolic pathways known as the classical (or neutral) and the alternative (acidic) pathway. In the classical pathway, cholesterol is metabolized to 7α-hydroxycholesterol by CYP7A1 and then to CA by CYP8A1 or to CDCA by CYP27A1. On the other hand, in the acid pathway, CYP27A1 converts cholesterol into 27-hydroxycholesterol which is then metabolized by CYP7B1 into CDCA. The entero-hepatic circulation of bile acids is mediated by several bile acid transporters in the liver and intestine and regulated by the FXR/SHP and FGF19/FGFR4 pathways. After their synthesis, primary bile acids are excreted into bile through the bile salt export pump (BSEP). b After secretion in the duodenum majority of BA are transported back to the liver through the portal blood. BAs are reabsorbed in the liver by NTCP. In the hepatocyte, other transporters including MRP2 on the canalicular membrane and MRP3/MRP4, OSTα/OSTβ on the basolateral membrane are also capable of BA transport into systemic circulation. c Finally, BAs are also filtered by the glomeruli and then reabsorbed in renal tubules, again limiting their renal loss. ASBT sodium-dependent bile acid transporter, BSEP bile salt export pump, FGF15 fibroblast growth factor 15, FGF-R4 FGF receptor 4, MDR2 multidrug resistance protein 2, MRP2/3/4 multidrug resistance-associated protein 2/3/4, NTCP sodium taurocholate co-transporting polypeptide, OSTα/β organic solute transporter α/β, SHP small heterodimer partner
Bile Acid Metabolism
Bile acids are the end-product of cholesterol metabolism generated in the liver by a chain of enzymatic reactions that involve the products of 17 genes organized in two main metabolic pathways, known as “classic” and “alternative” [2]. These liver pathways generate mainly two primary bile acids, i.e., cholic acid and chenodeoxycholic acid (CA and CDCA). In the classic pathway, the first and rate limiting step is the cholesterol-7α-hydroxylase (CYP7A1), an enzyme that converts cholesterol in 7α-OH-cholesterol. In the alternative pathway, the first step is represented by the C27 hydroxylation of cholesterol by the sterol 27-hydroxylase (CYP27A1) [2]. The classical pathway, in humans, accounts for ≈80% of total bile acid pool in physiological settings [2, 3]. The alternative pathway or “acidic,” because the C27 hydroxylation generates a COOH group, mainly converts cholesterol to CDCA [2, 3]. The main final products of the two pathways in the liver are 3α-7α di-hydroxylated cholesterol derivatives, i.e., CDCA, and 3α-7α-12α-tri-hydroxylated derivatives, i.e., CA. In hepatocytes, these primary bile acids are amidated (i.e., conjugated) with glycine (G) or taurine (T), giving rise to the bile salts (GCDCA and GCA, and TCDCA and TCA) [2, 3]. In addition, bile acids can be conjugated with the glucuronic acid by the uridine 5’ diphospho-glucoronosyltransferases (UGT1A1, 2B4 and 2B7), or sulfated by the sulfotransferases (SULT2A1 and SULT2A8) at positions C3 and C7 [2, 3]. In humans, the conjugation with G is prevalent and accounts for ≈ 90% of the bile acid pool. Conjugated bile acids are secreted in the intestine, becoming the substrate of an array of bacterial enzymes. These bio-transformations give rise to two secondary bile acids, i.e., lithocholic acid and deoxycholic acid (LCA and DCA). The “gateway reaction” of bile acid metabolism by intestinal bacteria is the enzymatic hydrolysis of the C-24N-acylamide bond operated by bile salt hydrolases (BSHs) which are found in all major bacterial phyla of the intestinal microbiota and some human gut archaea (Table 2 [4,5,6,7,8,9,10,11,12,13,14]). This first step is followed by the 7α-dehydroxylation of the OH in the C7 position, a reaction mediated by 7α-dehydroxylase expressing bacteria such as Clostridium and Eubacterium (Fig. 1). These reactions give rise to two main secondary bile acid, i.e., the 3α-mono-hydroxylated bile acids (i.e., LCA from CDCA), and 3α-12α-di-hydroxylated bile acids (i.e., DCA from CA) [2, 3, 5]. Additionally, the C7 β-epimerization of CDCA by Bacteroides, Clostridium, Escherichia, Eubacerium (and others) originates the 7β epimer of CDCA, i.e., the 3α,7β-dihydroxy-5β-cholanoic acid, known as ursodeoxycholic acid (UDCA) (Fig. 1 and Table 1 and 2) [15]. In addition to secondary bile acids, the intestinal microbiota generates 3, 7, and 12 oxo-bile acid analogues which contribute for a significant fraction (20–30%) of the bile acid metabolites produced by gut microbiota in the colon (Table 1). The bacterial genes involved in generation of oxo-bile acids are the 3α, 7α and 12α-hydroxysteroid dehydrogenases (HSDHs), which are mainly found in Clostridium clusters XIVa (i.e., C. scindens, C. hiranonis, and C. hylemonae), IV, and XI (Table 2) [5]. These oxo-bile acids are gaining growing attention because the ability of some 3-oxo-bile acids to bind to the retinoid-related orphan receptor (ROR)ɣt [16, 17]. In humans, the bile acid pool consists of CA (≈40%), CDCA (≈ 40%), DCA (≈ 20%), with a glycine over taurine conjugation ratio of 3–1 [18].
In rodents, there are additional bile acids, the α- and β-muricholic acids (MCA), generated in the liver from CDCA [19, 20]. These bile acids represent the product of specific murine gene, Cyp2c70, which is absent in human, and as such Cyp2c70−/− mice present a bile “humanized” bile acid profile [20,21,22]. Accordingly, the T conjugated of α- and β-MCA are primary bile acids in mice, while ω-MCA, generated from the two MCAs in the intestine, is a secondary bile acid. As such, there is almost no CDCA in mice, and while >90% of bile acids are T conjugated, the bile acid pool is made up by TCA (≈ 60%) and Tα-MCA and Tβ-MCA (≈ 40).
The large majority of bile acid species that reach the terminal ileum is reabsorbed by the intestinal epithelial cells (IEC) and transported back to the liver through the portal vein, completing a cycle in the so-called “entero-hepatic circulation” [2, 3]. A number of transporters and transcription factors are involved in the regulation of bile acid synthesis and metabolism in the liver and intestine and are described in Figs. 1 and 2 [2, 3, 15].

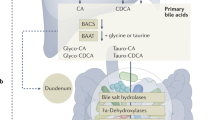
Intestinal bile acid metabolism. a The two primary bile acids, CA and CDCA, are then secreted into bile ducts and transported to the intestine and converted by the intestinal microbiota into secondary bile acids, respectively, DCA and LCA, or in the respective oxo- and di-oxo-derivatives. (b) In the gastrointestinal tract, the BAs are picked up by ASBT which transports them inside the enterocyte. BAs exit the enterocytes on the basolateral side via the IBABP/OSTα /OSTβ. Moreover, in the enterocite bile acids bind FXR which downregulates ASTB and upregulates the expression of IBABP and OSTα /OSTβ. Activation of FXR also induces the expression of SHP and the production of FGF15 which is then released into the portal circulation. In intestinal L cells, bile acids bind GPBAR1 which induces the production of GLP-1 which stimulates the secretion of insulin by the pancreas. (c) Expression levels of the GPBAR1, FXR, VDR, and RORC genes along the gastrointestinal tract extrapolated from https://www.proteinatlas.org/. Consensus Normalized eXpression (NX) levels created by combining the data from the three transcriptomics datasets (HPA, GTEx and FANTOM5) using the internal normalization pipeline. ASBT sodium-dependent bile acid transporter, FGF15 fibroblast growth factor 15, IBABP ileal bile acid-binding protein, MRP2/3 multidrug resistance-associated protein 2/3, OSTα/β organic solute transporter α/β, SHP small heterodimer partner
Bile Acid-Activated Receptors and Intestinal Immune Cells
Bile acids function as ligands for various receptors collectively known as “bile acid-activated receptors (BAR),” a family of cell membrane and nuclear receptors (Table 3) mainly expressed in the entero-hepatic system [15]. The best characterized of these receptors are the farnesoid-x-receptor (FXR, NR1H4), a nuclear transcription factor activated by primary bile acids, originally described in 1995 and deorphanized in 1999 [23,24,25], and the G-Protein Bile Acid-activated Receptor (GPBAR)-1, also known as TGR5, a seven-transmembrane G-protein-coupled receptor discovered in 2002 [26, 27] (Table 3). In addition, primary and secondary bile acids activate other nuclear receptors including the pregnane-x-receptor (PXR) [28], the constitutive androstane receptor (CAR) [29] and the vitamin D receptor (VDR) [111, 112] (Tables 8 and 9).
RORγt and IBD
RORγt is selectively expressed by Th17 lymphocytes and innate lymphoid cell group 3 (ILC3), acting as a critical transcription factor for Th17 cell differentiation in chronic inflammation and autoimmune diseases [113]. On the other hand, ROR-γt-dependent ILC3 provide a protective immunity [114,115,116,117]. Recently, S. Hang et al. [16] and X. Song et al. [17] have shown that oxo-bile acid derivatives, specifically the 3-oxo-LCA, can bind RORγt by acting as an inverse agonist. Both groups have shown that in a mouse models of colitis, the binding of RORγt decreases IL-17 production and Th17 cell number and attenuates intestinal inflammation. These data are consistent with finding that pharmacologic inhibition of ROR-γt provides therapeutic benefits in mouse models of intestinal inflammation and reduces the frequencies of Th17 cells but not ILC3s [118]. Studies in patients with IBD have shown that Th17 lymphocytes are involved in the pathogenesis of both CD and UC [119,120,121,122,123]. IL-17 expression in the mucosa and serum is increased in IBD patients and correlates with an increase in the expression of RORγt and the number of Th17 cells [124,125,126]. Despite that these data might suggest that transient inhibition of RORγt could be an effective in IBD and secukinumab a IL-17A blocking mAB has been shown beneficial in rheumatoid arthritis, a phase 2 clinical trial in active CD patients was interrupted for lack of efficacy and higher rate of adverse events in comparison with placebo [127]. Small molecules inverse agonist of RORγt may be, therefore, an alternative and effective approach to control Th17 immunity in IBD while boosting ILC3 function [128].
Vitamin D Receptor (VDR) and IBD
The VDR is a nuclear receptor activated by 1,25-dihydroxyvitamin D. The receptor is involved in the regulation of human metabolism, immunity, and cancer [129,130,131]. VDR is also activated by the secondary bile acid LCA and its metabolites (3-oxoLCA and iso-alloLCA) [17, 30]. VDR is mainly expressed in bone and intestine [132], but also in immune cells [133], and modulates both the innate and adaptative immune responses [129]. VDR activation blocks B cell proliferation and differentiation [134, 135], inhibits T cell proliferation [136], promotes a shift from a Th1 to a Th2 phenotype [137, 138], and drives T cell maturation facilitating the induction of T regulatory cells [139,140,141,142] and reducing the Th17 cell formation [143, 144]. In addition, it inhibits the production of inflammatory cytokines by monocytes such as IL-1, IL-6, IL-8, IL-12, and TNFα [145], and the differentiation and maturation of dendritic cells [146,147,148]. Results from preclinical models in mice have widely shown that vitamin D reduces the severity of colitis [149,150,151]. Vitamin D dietary deficiency exacerbates the symptoms in IL-10−/− mice in a model of enterocolitis, whereas dietary vitamin D supplementation improves diarrhea and prevents weight loss [152]. In addition to immune cell modulation, the VDR seems to be involved in regulating IEC homeostasis. Transgenic expression of human VDR in murine IEC is reported to protect mice from colitis by reducing IEC apoptosis and promoting the maintenance of intestinal mucosal barrier [153]. Accordingly, IEC-specific VDR KO mice show a more severe colitis and higher expression of TNF-α, IL-1β, and MCP-1 than wild-type mice [154, 155].
VDR signaling plays a beneficial role in clinical IBD [156, 157]. First of all, VDR polymorphisms (TaqI, BsmI, FokI, and ApaI) are associated with susceptibility to CD and UC [158]. In general, the distribution of VDR in intestinal tissue in patients with IBD correlates with mucosal inflammation. Low vitamin D levels in the plasma are associated with a poor prognosis, such as higher risk of surgery or increased risk of clinical relapse, in patients with UC [159]. In another study, Zator et al. proposed that low vitamin D levels may lead to earlier cessation of TNF-α therapy [160]. Furthermore, in a randomized double-blind placebo-controlled study, Jorgensen et al. showed that daily oral supplementation with 1200 IE vitamin D3 increased serum vitamin D levels and reduced the risk of relapse in CD patients from 29% to 13% (p = 0.06) [161]. However, the obtained result was not statistically significant; thus, further studies with larger populations are needed. In another clinical study, vitamin D (300,000 IU administered intramuscular) decreased the serum erythrocyte sedimentation rate and high-sensitivity C-reactive protein levels, in UC patients in remission after 90 days [162]. The beneficial effects of VDR signaling may be attributed to alterations in the resident microbiota. In two recent studies, the microbiota of CD and UC patients changed after early vitamin D administration [163, 164]. Other groups reported that vitamin D deficiency might worsen colitis through multiple effects including alterations of the gut microbiome [153, 165,166,167,168]. To date, however, we have only few available randomized double-blind, placebo-controlled studies investigating therapeutic effects of vitamin D in IBD.
PXR
PXR is a promiscuous receptor that, in addition to many endo- and xeno-compounds, accomodates LCA, thus functioning as LCA sensor [28].The human PXR is activated by the intestinal restricted antibiotic rifaximin, and its activation represses the intestinal immune response in a NF-KB-dependent manner [169, 170]. Additionally, probiotic metabolites activate intestinal PXR [171]. Several studies have investigated the genetic associations of the PXR gene polymorphism with IBD. Although a meta-analysis that included 6 studies suggested that 3 PXR SNPs (rs1523127, rs2276707, and rs6785049) had no obvious influence on the risk of IBD in Caucasians patients, further studies are needed to confirm the results [172]. PXR is expressed in human CD4+ and CD8+ T lymphocytes, CD19+ B lymphocytes, and CD14+ monocytes, and its activation in both mouse and human T cells inhibits T cell proliferation and CD25 and IFN-γ expression in vitro. In vivo PXR activation by pregnenolone 16α-carbonitrile (PCN a human PXR agonist) is protective against DSS-induced colitis due to the activation of phase II enzymes and cellular efflux transporters, such as GSTa1, MDR1a, and MRP2, which alleviates the expression of the pro-inflammatory cytokines IL-6, TNF-α, ΜCP-1, and IL-1β [169]. The protective effects are abrogated by PXR gene ablation. Mechanistically, PXR activation inhibits the activating effects of TNF-α on NF-κB. Rifaximin is an agonist for human, but not rodent, PXR. Using primary fetal human colon epithelial cells, Mencarelli et al. in this laboratory were able to show that rifaximin represses the expression of IL-6, TNF-α, and IL-8 mRNAs and promotes the expression of TGF-β by repressing lipopolysaccharide-induced NF-κB DNA-binding activity. These protective effects were abrogated PXR gene silencing in human macrophages [170]. As an antibiotic, rifaximin inhibits bacterial translocation, adhesion, and internalization [173]. Consistent with this view a randomized, double-blind placebo-controlled study has demonstrated that treating CD patients with a high dose formulation of rifaximin results in a higher 12-week clinical remission rate than placebo [174]. Rifaximin also effectively maintains remission in CD patients who had achieved remission with a standard therapy (100% of rifaximin-treated versus 87% of placebo-treated patients) [174].
Sphingosin1-Phosphate Receptor 2 (S1PR2)
The sphingosine1-phosphate (S1P) receptor 2 (S1PR2) is expressed in the ileum and colon and is activated by conjugated primary bile acids (GCA and TCA and GCDCA and TCDCA) [109]. Preliminary data suggest that S1PR2 deletion exacerbates intestinal inflammation caused by DSS in mice and that a S1P/S1PR2 pathway modulates MHC-II expression and regulates CD4+T-cell proliferation via the extracellular signal-regulated kinase (ERK) pathway [175, 176]. Zonulin-1 expression is increased by S1P analogue and decreased by S1PR2 antagonist. Several S1P modulators with differing selectivity toward S1P receptors have been advanced for clinical development for IBD. The S1PR1 antagonist ozanimod (RPC1063) has provided encouraging results in the Phase 2 TOUCHSTONE trial, and a Phase 3 trial in patients with moderate-to-severe UC is ongoing [177]. Etrasimod, a S1Pr1, S1pR3, and S1Pr5 antagonist, has also been investigated in patients with moderately to severely active UC. At the dose of 2 mg etrasimod was found more effective than placebo in producing clinical and endoscopic improvement, suggesting that further investigations are warranted [178]. Both ozanimod and etrasimod inhibit various S1PRs but not S1PR2; therefore, the role of S1PR2 in IBD remains to be proven.
Conclusions: Are Bile Acid-Based Therapies an Opportunity in IBD?
The data discussed so far illustrate that not only a dysregulation of bile acid signaling mediates the dysfunctional communication between the intestinal microbiota and immune system in IBD, but that bile acid-based therapies could by exploited for treating IBD. A number of the bile acid-activated receptors are altered in IBD patients, and restoring bile acid signaling might be beneficial in IBD as demonstrated for VDR and PXR. The two main bile acid receptors FXR and GPBAR1, however, have been poorly investigated for their therapeutic potential in this setting, despite the fact that a defective expression of FXR, gene and protein, increases the propensity to develop inflammation in mice and in humans. At the moment, there are no FXR agonists under development for IBD, but several non-selective and intestinal FXR selective agents are currently available. It could be anticipated, however, that there will be several drawbacks in using FXR ligands in treating IBD. First of all, FXR ligands cause side effects, the most common of which being itching. This side effect is dose dependent, and almost all currently available synthetic FXR ligands cause itching in a dose-dependent manner [179]. Additionally, FXR ligands exert profound effects on bile acid synthesis and cholesterol metabolism [109]. Because one of the main mechanisms involved in FXR-dependent inhibition of bile acid synthesis is the FGF15-/FGF-19-mediated repression of Cyp7A1, activation of intestinal FXR will result in liver repression of bile acid synthesis. The biological relevance of this effect in IBD patients remains unclear, but casts severe doubt that an FXR ligand could ever be developed for IBD.
In contrast, there is an increasing interest for GPBAR1 agonists. The expression of GPBAR1 is restricted to the intestine, and the receptor is not expressed by liver parenchymal cells. Data from preclinical models suggest that GPBAR1 ligand [102] exerts profound immune modulatory effects in rodent models of colitis. Potential side effects of a GPBAR1 ligand might include, again, itching since GPBAR1 (TGR5) activation has been associated with bile acid-induced pruritus in rodents, and its ablation attenuated pruritus caused by topical application of bile acid to the skin [103]. Because there are no GPBAR1 ligands currently under development, it is unclear whether or not the receptor mediates itching in humans. However, UDCA which we have shown to be a weak GPBAR1 ligand has been used to treat IBD and experimental data suggest a potential useful role for this agent in this setting [180,181,182,183,184,185]. Another potential complication of a GPBAR1 ligand could be diarrhea.
RORγt ligands hold potential in treating IBD, and works are in progress to identify RORγt reverse agonists (i.e., antagonists) to target intestinal inflammation.
Finally, there are several approaches that could be used for indirect modulation of intestinal FXR, GPBAR1, and RORγt by harnessing the intestinal microbiota by using probiotics or fecal microbial transplantation [6, 186,187,188,189,190,191]. Development of live bio-therapeutics engineered to produce ligands for beneficial receptors is another appealing possibility.
In conclusion, moving from the intestinal surface that promotes nutrient absorption, bile acids and their receptors have been revealed as an essential component of the chemical communications between the intestinal microbiota and the host. Altered bile acid signaling impacts on intestinal homeostasis and promotes the immune dysfunction seen in IBD, making bile acid receptors an interesting therapeutic target in IBD.
References
Mehrmal S, Uppal P, Nedley N, Giesey RL, Delost GR. The global, regional, and national burden of psoriasis in 195 countries and territories, 1990 to 2017: a systematic analysis from the Global Burden of Disease Study 2017. J Am Acad Dermatol. 1990. https://doi.org/10.1016/j.jaad.2020.04.139.
Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. https://doi.org/10.1194/jlr.R900010-JLR200.
Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem. 2003;72:137–174. https://doi.org/10.1146/annurev.biochem.72.121801.161712.
Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105:13580–13585. https://doi.org/10.1073/pnas.0804437105.
Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47:241–259. https://doi.org/10.1194/jlr.R500013-JLR200.
Long SL, Gahan CGM, Joyce SA. Interactions between gut bacteria and bile in health and disease. Mol Aspects Med. 2017;56:54–65. https://doi.org/10.1016/j.mam.2017.06.002.
Geng W, Lin J. Bacterial bile salt hydrolase: an intestinal microbiome target for enhanced animal health. Anim Health Res Rev. 2016;17:148–158. https://doi.org/10.1017/S1466252316000153.
Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes. 2016;7:22–39. https://doi.org/10.1080/19490976.2015.1127483.
Macdonald IA, Meier EC, Mahony DE, Costain GA. 3alpha-, 7alpha- and 12alpha-hydroxysteroid dehydrogenase activities from Clostridium perfringens. Biochim Biophys Acta. 1976;450:142–153. https://doi.org/10.1016/0005-2760(76)90086-2.
Doden H, Sallam LA, Devendran S, et al. Metabolism of oxo-bile acids and characterization of recombinant 12α-hydroxysteroid dehydrogenases from bile acid 7α-dehydroxylating human gut bacteria. Appl Environ Microbiol. 2018. https://doi.org/10.1128/AEM.00235-18.
Wells JE, Hylemon PB. Identification and characterization of a bile acid 7alpha-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7alpha-dehydroxylating strain isolated from human feces. Appl Environ Microbiol. 2000;66:1107–1113. https://doi.org/10.1128/aem.66.3.1107-1113.2000.
Hirano S, Nakama R, Tamaki M, Masuda N, Oda H. Isolation and characterization of thirteen intestinal microorganisms capable of 7 alpha-dehydroxylating bile acids. Appl Environ Microbiol. 1981;41:737–745. https://doi.org/10.1128/AEM.41.3.737-745.1981.
Doerner KC, Takamine F, LaVoie CP, Mallonee DH, Hylemon PB. Assessment of fecal bacteria with bile acid 7 alpha-dehydroxylating activity for the presence of bai-like genes. Appl Environ Microbiol. 1997;63:1185–1188. https://doi.org/10.1128/AEM.63.3.1185-1188.1997.
Gérard P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens. 2013;3:14–24. https://doi.org/10.3390/pathogens3010014.
Fiorucci S, Distrutti E. Chenodeoxycholic acid: an update on its therapeutic applications. Handb Exp Pharmacol. 2019;256:265–282. https://doi.org/10.1007/164_2019_226.
Hang S, Paik D, Yao L, et al. Bile acid metabolites control T. Nature. 2019;576:143–148. https://doi.org/10.1038/s41586-019-1785-z.
Song X, Sun X, Oh SF, et al. Microbial bile acid metabolites modulate gut RORγ. Nature. 2020;577:410–415. https://doi.org/10.1038/s41586-019-1865-0.
Chiang JY. Recent advances in understanding bile acid homeostasis. J Res. 2017;6:2029. https://doi.org/10.12688/f1000research.12449.1.
Chiang JY. Bile acid metabolism and signaling. Compr Physiol. 2013;3:1191–1212. https://doi.org/10.1002/cphy.c120023.
de Boer JF, Verkade E, Mulder NL, et al. A human-like bile acid pool induced by deletion of hepatic. J Lipid Res. 2020;61:291–305. https://doi.org/10.1194/jlr.RA119000243.
Straniero S, Laskar A, Savva C, Härdfeldt J, Angelin B, Rudling M. Of mice and men: murine bile acids explain species differences in the regulation of bile acid and cholesterol metabolism. J Lipid Res. 2020;61:480–491. https://doi.org/10.1194/jlr.RA119000307.
Takahashi S, Fukami T, Masuo Y, et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res. 2016;57:2130–2137. https://doi.org/10.1194/jlr.M071183.
Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284:1365–1368. https://doi.org/10.1126/science.284.5418.1365.
Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell. 1999;3:543–553. https://doi.org/10.1016/s1097-2765(00)80348-2.
Makishima M, Okamoto AY, Repa JJ, et al. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–1365. https://doi.org/10.1126/science.284.5418.1362.
Maruyama T, Miyamoto Y, Nakamura T, et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun. 2002;298:714–719. https://doi.org/10.1016/s0006-291x(02)02550-0.
Kawamata Y, Fujii R, Hosoya M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–9440. https://doi.org/10.1074/jbc.M209706200.
Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci USA. 2001;98:3369–3374. https://doi.org/10.1073/pnas.051551698.
Moore LB, Maglich JM, McKee DD, et al. Pregnane X receptor (PXR), constitutive androstane receptor (CAR), and benzoate X receptor (BXR) define three pharmacologically distinct classes of nuclear receptors. Mol Endocrinol. 2002;16:977–986. https://doi.org/10.1210/mend.16.5.0828.
Makishima M, Lu TT, **e W, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–1316. https://doi.org/10.1126/science.1070477.
Nagahashi M, Takabe K, Liu R, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. 2015;61:1216–1226. https://doi.org/10.1002/hep.27592.
De Marino S, Carino A, Masullo D, et al. Hyodeoxycholic acid derivatives as liver X receptor α and G-protein-coupled bile acid receptor agonists. Sci Rep. 2017;7:43290. https://doi.org/10.1038/srep43290.
Carino A, Biagioli M, Marchianò S, et al. Ursodeoxycholic acid is a GPBAR1 agonist and resets liver/intestinal FXR signaling in a model of diet-induced dysbiosis and NASH. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:1422–1437. https://doi.org/10.1016/j.bbalip.2019.07.006.
Sun L, **e C, Wang G, et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med. 2018;24:1919–1929. https://doi.org/10.1038/s41591-018-0222-4.
Jiang C, **e C, Lv Y, et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun. 2015;6:10166. https://doi.org/10.1038/ncomms10166.
Li F, Jiang C, Krausz KW, et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun. 2013;4:2384. https://doi.org/10.1038/ncomms3384.
Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. https://doi.org/10.1621/nrs.07003.
Cook DN, Kang HS, Jetten AM. retinoic acid-related orphan receptors (RORs) regulatory functions in immunity, development, circadian rhythm, and metabolism. Nucl Receptor Res. 2015. https://doi.org/10.11131/2015/101185.
Montaldo E, Juelke K, Romagnani C. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol. 2015;45:2171–2182. https://doi.org/10.1002/eji.201545598.
Eberl G, Marmon S, Sunshine MJ, Rennert PD, Choi Y, Littman DR. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat Immunol. 2004;5:64–73. https://doi.org/10.1038/ni1022.
Ivanov II, McKenzie BS, Zhou L, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. https://doi.org/10.1016/j.cell.2006.07.035.
Scoville SD, Freud AG, Caligiuri MA. Cellular pathways in the development of human and murine innate lymphoid cells. Curr Opin Immunol. 2019;56:100–106. https://doi.org/10.1016/j.coi.2018.11.003.
Fiorucci S, Biagioli M, Zampella A, Distrutti E. Bile acids activated receptors regulate innate immunity. Front Immunol. 2018;9:1853. https://doi.org/10.3389/fimmu.2018.01853.
Schote AB, Turner JD, Schiltz J, Muller CP. Nuclear receptors in human immune cells: expression and correlations. Mol Immunol. 2007;44:1436–1445. https://doi.org/10.1016/j.molimm.2006.04.021.
Lloyd-Price J, Arze C, Ananthakrishnan AN, et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 2019;569:655–662. https://doi.org/10.1038/s41586-019-1237-9.
Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci USA. 2007;104:13780–13785. https://doi.org/10.1073/pnas.0706625104.
Gevers D, Kugathasan S, Denson LA, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 2014;15:382–392. https://doi.org/10.1016/j.chom.2014.02.005.
Norman JM, Handley SA, Baldridge MT, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160:447–460. https://doi.org/10.1016/j.cell.2015.01.002.
Hoarau G, Mukherjee PK, Gower-Rousseau C, et al. Bacteriome and mycobiome interactions underscore microbial dysbiosis in familial Crohn’s disease. mBio. 2016.https://doi.org/10.1128/mBio.01250-16.
Zwolinska-Wcislo M, Brzozowski T, Budak A, et al. Effect of Candida colonization on human ulcerative colitis and the healing of inflammatory changes of the colon in the experimental model of colitis ulcerosa. J Physiol Pharmacol. 2009;60:107–118.
Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312:1355–1359. https://doi.org/10.1126/science.1124234.
Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. https://doi.org/10.1152/physrev.00045.2009.
Qin J, Li R, Raes J, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. https://doi.org/10.1038/nature08821.
Tap J, Mondot S, Levenez F, et al. Towards the human intestinal microbiota phylogenetic core. Environ Microbiol. 2009;11:2574–2584. https://doi.org/10.1111/j.1462-2920.2009.01982.x.
O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7:688–693. https://doi.org/10.1038/sj.embor.7400731.
Lederberg J. Infectious history. Science. 2000;288:287–293. https://doi.org/10.1126/science.288.5464.287.
Sender R, Fuchs S, Milo R. Are we really vastly outnumbered? Revisiting the ratio of bacterial to host cells in humans. Cell. 2016;164:337–340. https://doi.org/10.1016/j.cell.2016.01.013.
Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. https://doi.org/10.1038/nature11550.
Franzosa EA, Sirota-Madi A, Avila-Pacheco J, et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat Microbiol. 2019;4:293–305. https://doi.org/10.1038/s41564-018-0306-4.
Manichanh C, Rigottier-Gois L, Bonnaud E, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–211. https://doi.org/10.1136/gut.2005.073817.
Baumgart M, Dogan B, Rishniw M, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn’s disease involving the ileum. ISME J. 2007;1:403–418. https://doi.org/10.1038/ismej.2007.52.
Giaffer MH, Holdsworth CD, Duerden BI. The assessment of faecal flora in patients with inflammatory bowel disease by a simplified bacteriological technique. J Med Microbiol. 1991;35:238–243. https://doi.org/10.1099/00222615-35-4-238.
Mosca A, Leclerc M, Hugot JP. Gut microbiota diversity and human diseases: should we reintroduce key predators in our ecosystem? Front Microbiol. 2016;7:455. https://doi.org/10.3389/fmicb.2016.00455.
Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut. 2004;53:685–693. https://doi.org/10.1136/gut.2003.025403.
Gophna U, Sommerfeld K, Gophna S, Doolittle WF, van Veldhuyzen Zanten SJ. Differences between tissue-associated intestinal microfloras of patients with Crohn’s disease and ulcerative colitis. J Clin Microbiol. 2006;44:4136–4141. https://doi.org/10.1128/JCM.01004-06.
Atarashi K, Suda W, Luo C, et al. Ectopic colonization of oral bacteria in the intestine drives T. Science. 2017;358:359–365. https://doi.org/10.1126/science.aan4526.
Schmitz JM, Tonkonogy SL, Dogan B, et al. Murine adherent and invasive E. coli induces chronic inflammation and immune responses in the small and large intestines of monoassociated IL-10−/− mice independent of long polar fimbriae adhesin A. Inflamm Bowel Dis. 2019;25:875–885. https://doi.org/10.1093/ibd/izy386.
Darfeuille-Michaud A, Boudeau J, Bulois P, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology. 2004;127:412–421. https://doi.org/10.1053/j.gastro.2004.04.061.
Ohkusa T, Okayasu I, Ogihara T, Morita K, Ogawa M, Sato N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut. 2003;52:79–83. https://doi.org/10.1136/gut.52.1.79.
Imdad A, Nicholson MR, Tanner-Smith EE, et al. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst Rev. 2018;11:CD012774. https://doi.org/10.1002/14651858.CD012774.pub2.
Moayyedi P, Surette MG, Kim PT, et al. Fecal microbiota transplantation induces remission in patients with active ulcerative colitis in a randomized controlled trial. Gastroenterology. 2015;149:102–109. https://doi.org/10.1053/j.gastro.2015.04.001.
Paramsothy S, Kamm MA, Kaakoush NO, et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet. 2017;389:1218–1228. https://doi.org/10.1016/S0140-6736(17)30182-4.
Rossen NG, Fuentes S, van der Spek MJ, et al. Findings from a randomized controlled trial of fecal transplantation for patients with ulcerative colitis. Gastroenterology. 2015;149:110–188. https://doi.org/10.1053/j.gastro.2015.03.045.
van Nood E, Vrieze A, Nieuwdorp M, et al. Duodenal infusion of donor feces for recurrent Clostridium difficile. N Engl J Med. 2013;368:407–415. https://doi.org/10.1056/NEJMoa1205037.
Lavelle A, Sokol H. Gut microbiota-derived metabolites as key actors in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 2020;17:223–237. https://doi.org/10.1038/s41575-019-0258-z.
Vantrappen G, Ghoos Y, Rutgeerts P, Janssens J. Bile acid studies in uncomplicated Crohn’s disease. Gut. 1977;18:730–735. https://doi.org/10.1136/gut.18.9.730.
Rutgeerts P, Ghoos Y, Vantrappen G. Kinetics of primary bile acids in patients with non-operated Crohn’s disease. Eur J Clin Invest. 1982;12:135–143. https://doi.org/10.1111/j.1365-2362.1982.tb00950.x.
Cummings JH, James WP, Wiggins HS. Role of the colon in ileal-resection diarrhoea. Lancet. 1973;1:344–347. https://doi.org/10.1016/s0140-6736(73)90131-1.
Mekhjian HS, Phillips SF, Hofmann AF. Colonic absorption of unconjugated bile acids: perfusion studies in man. Dig Dis Sci. 1979;24:545–550. https://doi.org/10.1007/BF01489324.
Midtvedt T, Norman A. Parameters in 7-alpha-dehydroxylation of bile acids by anaerobic lactobacilli. Acta Pathol Microbiol Scand. 1968;72:313–329. https://doi.org/10.1111/j.1699-0463.1968.tb01345.x.
Kruis W, Kalek HD, Stellaard F, Paumgartner G. Altered fecal bile acid pattern in patients with inflammatory bowel disease. Digestion. 1986;35:189–198. https://doi.org/10.1159/000199367.
Macdonald IA, Singh G, Mahony DE, Meier CE. Effect of pH on bile salt degradation by mixed fecal cultures. Steroids. 1978;32:245–256. https://doi.org/10.1016/0039-128x(78)90009-0.
Aries V, Hill MJ. Degradation of steroids by intestinal bacteria. I. Deconjugation of bile salts. Biochim Biophys Acta. 1970;202:526–534. https://doi.org/10.1016/0005-2760(70)90123-2.
Duboc H, Rajca S, Rainteau D, et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut. 2013;62:531–539. https://doi.org/10.1136/gutjnl-2012-302578.
Ding L, Yang L, Wang Z, Huang W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm Sin B. 2015;5:135–144. https://doi.org/10.1016/j.apsb.2015.01.004.
Plass JR, Mol O, Heegsma J, et al. Farnesoid X receptor and bile salts are involved in transcriptional regulation of the gene encoding the human bile salt export pump. Hepatology. 2002;35:589–596. https://doi.org/10.1053/jhep.2002.31724.
Kok T, Hulzebos CV, Wolters H, et al. Enterohepatic circulation of bile salts in farnesoid X receptor-deficient mice: efficient intestinal bile salt absorption in the absence of ileal bile acid-binding protein. J Biol Chem. 2003;278:41930–41937. https://doi.org/10.1074/jbc.M306309200.
Gadaleta RM, Garcia-Irigoyen O, Cariello M, et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBioMedicine. 2020;54:102719. https://doi.org/10.1016/j.ebiom.2020.102719.
Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol. 2009;183:6251–6261. https://doi.org/10.4049/jimmunol.0803978.
Renga B, Mencarelli A, Cipriani S, et al. The bile acid sensor FXR is required for immune-regulatory activities of TLR-9 in intestinal inflammation. PLoS ONE. 2013;8:e54472. https://doi.org/10.1371/journal.pone.0054472.
Goodwin B, Jones SA, Price RR, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. https://doi.org/10.1016/s1097-2765(00)00051-4.
Inagaki T, Moschetta A, Lee YK, et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA. 2006;103:3920–3925. https://doi.org/10.1073/pnas.0509592103.
Gadaleta RM, Oldenburg B, Willemsen EC, et al. Activation of bile salt nuclear receptor FXR is repressed by pro-inflammatory cytokines activating NF-κB signaling in the intestine. Biochim Biophys Acta. 2011;1812:851–858. https://doi.org/10.1016/j.bbadis.2011.04.005.
Hao H, Cao L, Jiang C, et al. Farnesoid X receptor regulation of the NLRP3 inflammasome underlies cholestasis-associated sepsis. Cell Metab. 2017;25:856–867. https://doi.org/10.1016/j.cmet.2017.03.007.
Torres J, Bao X, Iuga AC, et al. Farnesoid X receptor expression is decreased in colonic mucosa of patients with primary sclerosing cholangitis and colitis-associated neoplasia. Inflamm Bowel Dis. 2013;19:275–282. https://doi.org/10.1097/MIB.0b013e318286ff2e.
Wilson A, Almousa A, Teft WA, Kim RB. Attenuation of bile acid-mediated FXR and PXR activation in patients with Crohn’s disease. Sci Rep. 2020;10:1866. https://doi.org/10.1038/s41598-020-58644-w.
Gadaleta RM, van Erpecum KJ, Oldenburg B, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut. 2011;60:463–472. https://doi.org/10.1136/gut.2010.212159.
Nijmeijer RM, Gadaleta RM, van Mil SW, et al. Farnesoid X receptor (FXR) activation and FXR genetic variation in inflammatory bowel disease. PLoS ONE. 2011;6:e23745. https://doi.org/10.1371/journal.pone.0023745.
Attinkara R, Mwinyi J, Truninger K, et al. Association of genetic variation in the NR1H4 gene, encoding the nuclear bile acid receptor FXR, with inflammatory bowel disease. BMC Res Notes. 2012;5:461. https://doi.org/10.1186/1756-0500-5-461.
Wilson A, Wang Q, Almousa AA, et al. Genetic variation in the farnesoid X-receptor predicts Crohn’s disease severity in female patients. Sci Rep. 2020;10:11725. https://doi.org/10.1038/s41598-020-68686-9.
Cipriani S, Mencarelli A, Chini MG, et al. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE. 2011;6:e25637. https://doi.org/10.1371/journal.pone.0025637.
Biagioli M, Carino A, Cipriani S, et al. The bile acid receptor GPBAR1 regulates the M1/M2 phenotype of intestinal macrophages and activation of GPBAR1 rescues mice from murine colitis. J Immunol. 2017;199:718–733. https://doi.org/10.4049/jimmunol.1700183.
Alemi F, Poole DP, Chiu J, et al. The receptor TGR5 mediates the prokinetic actions of intestinal bile acids and is required for normal defecation in mice. Gastroenterology. 2013;144:145–154. https://doi.org/10.1053/j.gastro.2012.09.055.
Castro J, Harrington AM, Lieu T, et al. Activation of pruritogenic TGR5, MrgprA3, and MrgprC11 on colon-innervating afferents induces visceral hypersensitivity. JCI Insight. 2019. https://doi.org/10.1172/jci.insight.131712.
Poole DP, Godfrey C, Cattaruzza F, et al. Expression and function of the bile acid receptor GpBAR1 (TGR5) in the murine enteric nervous system. Neurogastroenterol Motil. 2010;22:814–825. https://doi.org/10.1111/j.1365-2982.2010.01487.x.
Hov JR, Keitel V, Laerdahl JK, et al. Mutational characterization of the bile acid receptor TGR5 in primary sclerosing cholangitis. PLoS ONE. 2010;5:e12403. https://doi.org/10.1371/journal.pone.0012403.
Yusta B, Holland D, Koehler JA, et al. ErbB signaling is required for the proliferative actions of GLP-2 in the murine gut. Gastroenterology. 2009;137:986–996. https://doi.org/10.1053/j.gastro.2009.05.057.
Reich M, Deutschmann K, Sommerfeld A, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65:487–501. https://doi.org/10.1136/gutjnl-2015-309458.
Fiorucci S, Distrutti E. The pharmacology of bile acids and their receptors. Handb Exp Pharmacol. 2019;256:3–18. https://doi.org/10.1007/164_2019_238.
Keitel V, Stindt J, Häussinger D. Bile acid-activated receptors: GPBAR1 (TGR5) and other G protein-coupled receptors. Handb Exp Pharmacol. 2019;256:19–49. https://doi.org/10.1007/164_2019_230.
Biagioli M, Carino A, Fiorucci C, et al. GPBAR1 functions as gatekeeper for liver NKT cells and provides counterregulatory signals in mouse models of immune-mediated hepatitis. Cell Mol Gastroenterol Hepatol. 2019;8:447–473. https://doi.org/10.1016/j.jcmgh.2019.06.003.
Biagioli M, Carino A, Fiorucci C, et al. The bile acid receptor GPBAR1 modulates CCL2/CCR2 signaling at the liver sinusoidal/macrophage interface and reverses acetaminophen-induced liver toxicity. J Immunol. 2020;204:2535–2551. https://doi.org/10.4049/jimmunol.1901427.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. https://doi.org/10.1146/annurev.immunol.021908.132710.
Eberl G, Littman DR. The role of the nuclear hormone receptor RORgammat in the development of lymph nodes and Peyer’s patches. Immunol Rev. 2003;195:81–90. https://doi.org/10.1034/j.1600-065x.2003.00074.x.
Sawa S, Lochner M, Satoh-Takayama N, et al. RORγt+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat Immunol. 2011;12:320–326. https://doi.org/10.1038/ni.2002.
Mortha A, Chudnovskiy A, Hashimoto D, et al. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science. 2014;343:1249288. https://doi.org/10.1126/science.1249288.
Sonnenberg GF, Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. 2015;21:698–708. https://doi.org/10.1038/nm.3892.
Withers DR, Hepworth MR, Wang X, et al. Transient inhibition of ROR-γt therapeutically limits intestinal inflammation by reducing TH17 cells and preserving group 3 innate lymphoid cells. Nat Med. 2016;22:319–323. https://doi.org/10.1038/nm.4046.
Lee SH, Kwon JE, Cho ML. Immunological pathogenesis of inflammatory bowel disease. Intest Res. 2018;16:26–42. https://doi.org/10.5217/ir.2018.16.1.26.
Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58:1152–1167. https://doi.org/10.1136/gut.2008.163667.
Imam T, Park S, Kaplan MH, Olson MR. Effector T helper cell subsets in inflammatory bowel diseases. Front Immunol. 2018;9:1212. https://doi.org/10.3389/fimmu.2018.01212.
Gálvez J. Role of Th17 cells in the pathogenesis of human IBD. ISRN Inflamm. 2014;2014:928461. https://doi.org/10.1155/2014/928461.
Klepsch V, Moschen AR, Tilg H, Baier G, Hermann-Kleiter N. Nuclear receptors regulate intestinal inflammation in the context of IBD. Front Immunol. 2019;10:1070. https://doi.org/10.3389/fimmu.2019.01070.
Hou G, Bishu S. Th17 cells in inflammatory bowel disease: an update for the clinician. Inflamm Bowel Dis. 2020;26:653–661. https://doi.org/10.1093/ibd/izz316.
Jiang W, Su J, Zhang X, et al. Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm Res. 2014;63:943–950. https://doi.org/10.1007/s00011-014-0768-7.
Fu**o S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. https://doi.org/10.1136/gut.52.1.65.
Hueber W, Sands BE, Lewitzky S, et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. https://doi.org/10.1136/gutjnl-2011-301668.
Bassolas-Molina H, Raymond E, Labadia M, et al. An RORγt oral inhibitor modulates IL-17 responses in peripheral blood and intestinal mucosa of Crohn’s disease patients. Front Immunol. 2018;9:2307. https://doi.org/10.3389/fimmu.2018.02307.
Aranow C. Vitamin D and the immune system. J Investig Med. 2011;59:881–886. https://doi.org/10.2310/JIM.0b013e31821b8755.
Bouillon R, Carmeliet G, Verlinden L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–776. https://doi.org/10.1210/er.2008-0004.
Bandera Merchan B, Morcillo S, Martin-Nuñez G, Tinahones FJ, Macías-González M. The role of vitamin D and VDR in carcinogenesis: through epidemiology and basic sciences. J Steroid Biochem Mol Biol. 2017;167:203–218. https://doi.org/10.1016/j.jsbmb.2016.11.020.
Wang Y, Zhu J, DeLuca HF. Where is the vitamin D receptor? Arch Biochem Biophys. 2012;523:123–133. https://doi.org/10.1016/j.abb.2012.04.001.
Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. 2009;94:26–34. https://doi.org/10.1210/jc.2008-1454.
Lemire JM, Adams JS, Sakai R, Jordan SC. 1 alpha,25-dihydroxyvitamin D3 suppresses proliferation and immunoglobulin production by normal human peripheral blood mononuclear cells. J Clin Invest. 1984;74:657–661. https://doi.org/10.1172/JCI111465.
Chen S, Sims GP, Chen XX, Gu YY, Lipsky PE. Modulatory effects of 1,25-dihydroxyvitamin D3 on human B cell differentiation. J Immunol. 2007;179:1634–1647. https://doi.org/10.4049/jimmunol.179.3.1634.
Bhalla AK, Amento EP, Serog B, Glimcher LH. 1,25-Dihydroxyvitamin D3 inhibits antigen-induced T cell activation. J Immunol. 1984;133:1748–1754.
Mattner F, Smiroldo S, Galbiati F, et al. Inhibition of Th1 development and treatment of chronic-relapsing experimental allergic encephalomyelitis by a non-hypercalcemic analogue of 1,25-dihydroxyvitamin D(3). Eur J Immunol. 2000;30:498–508. https://doi.org/10.1002/1521-4141(200002)30:2%3c498:AID-IMMU498%3e3.0.CO;2-Q.
Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HF, O’Garra A. 1alpha,25-Dihydroxyvitamin d3 has a direct effect on naive CD4(+) T cells to enhance the development of Th2 cells. J Immunol. 2001;167:4974–4980. https://doi.org/10.4049/jimmunol.167.9.4974.
Gregori S, Casorati M, Amuchastegui S, Smiroldo S, Davalli AM, Adorini L. Regulatory T cells induced by 1 alpha,25-dihydroxyvitamin D3 and mycophenolate mofetil treatment mediate transplantation tolerance. J Immunol. 2001;167:1945–1953. https://doi.org/10.4049/jimmunol.167.4.1945.
Barrat FJ, Cua DJ, Boonstra A, et al. In vitro generation of interleukin 10-producing regulatory CD4(+) T cells is induced by immunosuppressive drugs and inhibited by T helper type 1 (Th1)- and Th2-inducing cytokines. J Exp Med. 2002;195:603–616. https://doi.org/10.1084/jem.20011629.
Gorman S, Kuritzky LA, Judge MA, et al. Topically applied 1,25-dihydroxyvitamin D3 enhances the suppressive activity of CD4+CD25+ cells in the draining lymph nodes. J Immunol. 2007;179:6273–6283. https://doi.org/10.4049/jimmunol.179.9.6273.
Penna G, Roncari A, Amuchastegui S, et al. Expression of the inhibitory receptor ILT3 on dendritic cells is dispensable for induction of CD4+Foxp3+ regulatory T cells by 1,25-dihydroxyvitamin D3. Blood. 2005;106:3490–3497. https://doi.org/10.1182/blood-2005-05-2044.
Tang J, Zhou R, Luger D, et al. Calcitriol suppresses antiretinal autoimmunity through inhibitory effects on the Th17 effector response. J Immunol. 2009;182:4624–4632. https://doi.org/10.4049/jimmunol.0801543.
Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther. 2008;324:23–33. https://doi.org/10.1124/jpet.107.127209.
Almerighi C, Sinistro A, Cavazza A, Ciaprini C, Rocchi G, Bergamini A. 1Alpha,25-dihydroxyvitamin D3 inhibits CD40L-induced pro-inflammatory and immunomodulatory activity in human monocytes. Cytokine. 2009;45:190–197. https://doi.org/10.1016/j.cyto.2008.12.009.
Piemonti L, Monti P, Sironi M, et al. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol. 2000;164:4443–4451. https://doi.org/10.4049/jimmunol.164.9.4443.
Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1alpha,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A. 2001;98:6800–6805. https://doi.org/10.1073/pnas.121172198.
Széles L, Keresztes G, Töröcsik D, et al. 1,25-dihydroxyvitamin D3 is an autonomous regulator of the transcriptional changes leading to a tolerogenic dendritic cell phenotype. J Immunol. 2009;182:2074–2083. https://doi.org/10.4049/jimmunol.0803345.
Li YC, Chen Y, Du J. Critical roles of intestinal epithelial vitamin D receptor signaling in controlling gut mucosal inflammation. J Steroid Biochem Mol Biol. 2015;148:179–183. https://doi.org/10.1016/j.jsbmb.2015.01.011.
Zhu W, Yan J, Zhi C, Zhou Q, Yuan X. 1,25(OH). Gut Pathog. 2019;11:8. https://doi.org/10.1186/s13099-019-0291-z.
Daniel C, Radeke HH, Sartory NA, et al. The new low calcemic vitamin D analog 22-ene-25-oxa-vitamin D prominently ameliorates T helper cell type 1-mediated colitis in mice. J Pharmacol Exp Ther. 2006;319:622–631. https://doi.org/10.1124/jpet.106.107599.
Cantorna MT, Munsick C, Bemiss C, Mahon BD. 1,25-Dihydroxycholecalciferol prevents and ameliorates symptoms of experimental murine inflammatory bowel disease. J Nutr. 2000;130:2648–2652. https://doi.org/10.1093/jn/130.11.2648.
Liu W, Chen Y, Golan MA, et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J Clin Invest. 2013;123:3983–3996. https://doi.org/10.1172/JCI65842.
Kong J, Zhang Z, Musch MW, et al. Novel role of the vitamin D receptor in maintaining the integrity of the intestinal mucosal barrier. Am J Physiol Gastrointest Liver Physiol. 2008;294:G208–G216. https://doi.org/10.1152/ajpgi.00398.2007.
Kim JH, Yamaori S, Tanabe T, et al. Implication of intestinal VDR deficiency in inflammatory bowel disease. Biochim Biophys Acta. 2013;1830:2118–2128. https://doi.org/10.1016/j.bbagen.2012.09.020.
Gubatan J, Chou ND, Nielsen OH, Moss AC. Systematic review with meta-analysis: association of vitamin D status with clinical outcomes in adult patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2019;50:1146–1158. https://doi.org/10.1111/apt.15506.
van der Post S, Jabbar KS, Birchenough G, et al. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut. 2019;68:2142–2151. https://doi.org/10.1136/gutjnl-2018-317571.
Xue LN, Xu KQ, Zhang W, Wang Q, Wu J, Wang XY. Associations between vitamin D receptor polymorphisms and susceptibility to ulcerative colitis and Crohn’s disease: a meta-analysis. Inflamm Bowel Dis. 2013;19:54–60. https://doi.org/10.1002/ibd.22966.
Gubatan J, Mitsuhashi S, Zenlea T, Rosenberg L, Robson S, Moss AC. Low Serum Vitamin D During Remission Increases Risk of Clinical Relapse in Patients With Ulcerative Colitis. Clin Gastroenterol Hepatol. 2017;15:240–246. https://doi.org/10.1016/j.cgh.2016.05.035.
Zator ZA, Cantu SM, Konijeti GG, et al. Pretreatment 25-hydroxyvitamin D levels and durability of anti-tumor necrosis factor-α therapy in inflammatory bowel diseases. JPEN J Parenter Enteral Nutr. 2014;38:385–391. https://doi.org/10.1177/0148607113504002.
Jørgensen SP, Agnholt J, Glerup H, et al. Clinical trial: vitamin D3 treatment in Crohn’s disease - a randomized double-blind placebo-controlled study. Aliment Pharmacol Ther. 2010;32:377–383. https://doi.org/10.1111/j.1365-2036.2010.04355.x.
Sharifi A, Hosseinzadeh-Attar MJ, Vahedi H, Nedjat S. A randomized controlled trial on the effect of vitamin D3 on inflammation and cathelicidin gene expression in ulcerative colitis patients. Saudi J Gastroenterol. 2016;22:316–323. https://doi.org/10.4103/1319-3767.187606.
Schäffler H, Herlemann DP, Klinitzke P, et al. Vitamin D administration leads to a shift of the intestinal bacterial composition in Crohn’s disease patients, but not in healthy controls. J Dig Dis. 2018;19:225–234. https://doi.org/10.1111/1751-2980.12591.
Garg M, Hendy P, Ding JN, Shaw S, Hold G, Hart A. The effect of vitamin D on intestinal inflammation and faecal microbiota in patients with ulcerative colitis. J Crohns Colitis. 2018;12:963–972. https://doi.org/10.1093/ecco-jcc/jjy052.
Sun J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy. 2016;12:1057–1058. https://doi.org/10.1080/15548627.2015.1072670.
Wu S, Zhang YG, Lu R, et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut. 2015;64:1082–1094. https://doi.org/10.1136/gutjnl-2014-307436.
Garg M, Rosella O, Lubel JS, Gibson PR. Association of circulating vitamin D concentrations with intestinal but not systemic inflammation in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2634–2643. https://doi.org/10.1097/01.MIB.0000436957.77533.b2.
** D, Wu S, Zhang YG, et al. Lack of Vitamin D Receptor Causes Dysbiosis and Changes the Functions of the Murine Intestinal Microbiome. Clin Ther. 2015;37:996–1009. https://doi.org/10.1016/j.clinthera.2015.04.004.
Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1114–G1122. https://doi.org/10.1152/ajpgi.00528.2006.
Mencarelli A, Migliorati M, Barbanti M, et al. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem Pharmacol. 2010;80:1700–1707. https://doi.org/10.1016/j.bcp.2010.08.022.
Mencarelli A, Distrutti E, Renga B, et al. Probiotics modulate intestinal expression of nuclear receptor and provide counter-regulatory signals to inflammation-driven adipose tissue activation. PLoS ONE. 2011;6:e22978. https://doi.org/10.1371/journal.pone.0022978.
Guo X, Yan M. Pregnane X receptor polymorphisms and risk of inflammatory bowel disease: a meta-analysis. Immunol Invest. 2017;46:566–576. https://doi.org/10.1080/08820139.2017.1322101.
Mencarelli A, Renga B, Palladino G, et al. Inhibition of NF-κB by a PXR-dependent pathway mediates counter-regulatory activities of rifaximin on innate immunity in intestinal epithelial cells. Eur J Pharmacol. 2011;668:317–324. https://doi.org/10.1016/j.ejphar.2011.06.058.
Prantera C, Lochs H, Grimaldi M, et al. Rifaximin-extended intestinal release induces remission in patients with moderately active Crohn’s disease. Gastroenterology. 2012;142:473–481. https://doi.org/10.1053/j.gastro.2011.11.032.
Chen T, Lin R, ** S, et al. The sphingosine-1-phosphate/sphingosine-1-phosphate receptor 2 Axis in intestinal epithelial cells regulates intestinal barrier function during intestinal epithelial cells-CD4+T-cell interactions. Cell Physiol Biochem. 2018;48:1188–1200. https://doi.org/10.1159/000491985.
Kwong EK, Zhou H. Sphingosine-1-phosphate signaling and the gut-liver axis in liver diseases. Liver Res. 2019;3:19–24. https://doi.org/10.1016/j.livres.2019.02.003.
Sandborn WJ, Feagan BG, Wolf DC, et al. Ozanimod Induction and Maintenance Treatment for Ulcerative Colitis. N Engl J Med. 2016;374:1754–1762. https://doi.org/10.1056/NEJMoa1513248.
Sandborn WJ, Peyrin-Biroulet L, Zhang J, et al. Efficacy and safety of etrasimod in a phase 2 randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158:550–561. https://doi.org/10.1053/j.gastro.2019.10.035.
Carino A, Biagioli M, Marchianò S, et al. Opposite effects of the FXR agonist obeticholic acid on Mafg and Nrf2 mediate the development of acute liver injury in rodent models of cholestasis. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865:158733. https://doi.org/10.1016/j.bbalip.2020.158733.
Laukens D, Devisscher L, Van den Bossche L, et al. Tauroursodeoxycholic acid inhibits experimental colitis by preventing early intestinal epithelial cell death. Lab Invest. 2014;94:1419–1430. https://doi.org/10.1038/labinvest.2014.117.
Martínez-Moya P, Romero-Calvo I, Requena P, et al. Dose-dependent antiinflammatory effect of ursodeoxycholic acid in experimental colitis. Int Immunopharmacol. 2013;15:372–380. https://doi.org/10.1016/j.intimp.2012.11.017.
Van den Bossche L, Borsboom D, Devriese S, et al. Tauroursodeoxycholic acid protects bile acid homeostasis under inflammatory conditions and dampens Crohn’s disease-like ileitis. Lab Invest. 2017;97:519–529. https://doi.org/10.1038/labinvest.2017.6.
Yang Y, He J, Suo Y, et al. Tauroursodeoxycholate improves 2,4,6-trinitrobenzenesulfonic acid-induced experimental acute ulcerative colitis in mice. Int Immunopharmacol. 2016;36:271–276. https://doi.org/10.1016/j.intimp.2016.04.037.
O’Dwyer AM, Lajczak NK, Keyes JA, Ward JB, Greene CM, Keely SJ. Ursodeoxycholic acid inhibits TNFα-induced IL-8 release from monocytes. Am J Physiol Gastrointest Liver Physiol. 2016;311:G334–G341. https://doi.org/10.1152/ajpgi.00406.2015.
Ward JBJ, Lajczak NK, Kelly OB, et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am J Physiol Gastrointest Liver Physiol. 2017;312:G550–G558. https://doi.org/10.1152/ajpgi.00256.2016.
Foley MH, O’Flaherty S, Barrangou R, Theriot CM. Bile salt hydrolases: gatekeepers of bile acid metabolism and host-microbiome crosstalk in the gastrointestinal tract. PLoS Pathog. 2019;15:e1007581. https://doi.org/10.1371/journal.ppat.1007581.
Ahmadi S, Wang S, Nagpal R, et al. A human-origin probiotic cocktail ameliorates aging-related leaky gut and inflammation via modulating the microbiota/taurine/tight junction axis. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.132055.
Joyce SA, Gahan CG. Bile acid modifications at the microbe-host interface: potential for nutraceutical and pharmaceutical interventions in host health. Annu Rev Food Sci Technol. 2016;7:313–333. https://doi.org/10.1146/annurev-food-041715-033159.
Ogilvie LA, Jones BV. Dysbiosis modulates capacity for bile acid modification in the gut microbiomes of patients with inflammatory bowel disease: a mechanism and marker of disease? Gut. 2012;61:1642–1643. https://doi.org/10.1136/gutjnl-2012-302137.
Joyce SA, Shanahan F, Hill C, Gahan CG. Bacterial bile salt hydrolase in host metabolism: potential for influencing gastrointestinal microbe-host crosstalk. Gut Microbes. 2014;5:669–674. https://doi.org/10.4161/19490976.2014.969986.
Buffie CG, Bucci V, Stein RR, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517:205–208. https://doi.org/10.1038/nature13828.
Funding
Open access funding provided by Università degli Studi di Perugia within the CRUI-CARE Agreement.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Prof. Stefano Fiorucci, is listed as an inventor of some of the compounds mentioned in this paper: INT767 (Intercept Pharmaceuticals), BAR501 and BAR502 (Bar Pharmaceuticals) and has received research grants from BAR Pharamceuticals. The other authors do not have any conflict of interest to be disclosed.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Fiorucci, S., Carino, A., Baldoni, M. et al. Bile Acid Signaling in Inflammatory Bowel Diseases. Dig Dis Sci 66, 674–693 (2021). https://doi.org/10.1007/s10620-020-06715-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10620-020-06715-3