
Abstract.
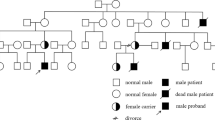
The mouse scurfy gene, Foxp3, and its human orthologue, FOXP3, which maps to Xp11.23–Xq13.3, were recently identified by positional cloning. Point mutations and microdeletions of the FOXP3 gene were found in the affected members of eight of nine families with IPEX (immune dysfunction, polyendocrinopathy, enteropathy, X-linked; OMIM 304930). We evaluated a pedigree with clinically typical IPEX in which mutations of the coding exons of FOXP3 were not detected. Our reevaluation of this pedigree identified an A→G transition within the first polyadenylation signal (AAUAAA→AAUGAA) after the stop codon. The next polyadenylation signal is not encountered for a further 5.1 kb. This transition was not detected in over 212 normal individuals (~318 X chromosomes), excluding the possibility of a rare polymorphism. We suggest that this mutation is causal of IPEX in this family by a mechanism of nonspecific degradation of the FOXP3 gene message.
Similar content being viewed by others

Author information
Authors and Affiliations
Additional information
Electronic Publication
Rights and permissions
About this article
Cite this article
Bennett, C.L., Brunkow, M.E., Ramsdell, F. et al. A rare polyadenylation signal mutation of the FOXP3 gene (AAUAAA→AAUGAA) leads to the IPEX syndrome. Immunogenetics 53, 435–439 (2001). https://doi.org/10.1007/s002510100358
Received:
Revised:
Issue Date:
DOI: https://doi.org/10.1007/s002510100358