
Abstract
Beta cells are primarily defined by their ability to produce insulin and secrete it in response to appropriate stimuli. It has been known for some time, however, that beta cells are not functionally identical to each other and that the rates of insulin synthesis and release differ from cell to cell, although the functional significance of this variability remains unclear. Recent studies have used heterogeneous gene expression to isolate and evaluate different subpopulations of beta cells and to demonstrate alterations in these subpopulations in diabetes. In the last few years, novel technologies have emerged that permit the detailed evaluation of the proteome (e.g. time-of-flight mass spectroscopy, [CyTOF]) and transcriptome (e.g. massively parallel RNA sequencing) at the single-cell level, and tools for single beta cell metabolomics and epigenomics are quickly maturing. The first wave of single beta cell proteome and transcriptome studies were published in 2016, giving a glimpse into the power, but also the limitations, of these approaches. Despite this progress, it remains unclear if the observed heterogeneity of beta cells represents stable, distinct beta cell types or, alternatively, highly dynamic beta cell states. Here we provide a concise overview of recent developments in the emerging field of beta cell heterogeneity and the implications for our understanding of beta cell biology and pathology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Adult pancreatic beta cells share the unique fundamental trait of expressing insulin and secreting it in response to relevant stimuli. When beta cells have been studied in bulk, either isolated or in situ, the primary regulatory factor for insulin secretion is glucose, both stimulating secretion when levels are high and suppressing secretion when they are low. However, other stimuli, such as amino acids, fatty acids, hormones and neuronal signals, also play important roles in determining the net secretion of insulin by a population of beta cells. Do all beta cells respond equally to all of these physiological signals, or are there subpopulations that respond differentially to specific signals? There is growing evidence (recently reviewed in detail by Roscioni et al [1]) to suggest that not all beta cells are functionally identical. If correct, this raises additional questions related to the physiological significance of the relative prevalence of functionally different beta cell subpopulations and the degree of plasticity of individual beta cells; questions that are not likely to be merely philosophical or trivial, since the answers may provide insight into the pathophysiology of beta cell disease, particularly type 1 and type 2 diabetes, and may identify novel pathogenic pathways amenable to intervention.
Evidence of beta cell structural heterogeneity dates back to the 1960s [2], whereas functional heterogeneity was first described in the mid 1980s, when Salomon and Meda first observed secretory heterogeneity using the reverse haemolytic plaque assay on individual rat beta cells [3]. This observation was subsequently confirmed and further investigated by both Bosco and Meda, and Pipeleers and colleagues [4,5,6,7]. These studies evaluated islets and beta cells isolated from the entire pancreas, with little consideration for their location within the pancreas or islet composition. Regional heterogeneity in rodent islets has been documented [8] and could result in functional differences in beta cells isolated from different regions of the pancreas. In contrast to the situation in rodents, human islet composition and function appear to be similar in all regions of the pancreas, although islet density is higher in the tail compared with the head or body [9]. Interestingly, in human type 1 diabetes, the severity of beta cell loss is greater in the tail of the pancreas compared with the head [10], whereas, in type 2 diabetes, beta cell loss is observed mostly in the head of the pancreas [9], suggesting functional differences between islets from different regions of the pancreas.
Neither the molecular mechanisms that drive this heterogeneity in rodent and human beta cells, nor its physiological relevance are known. However, recent technological advances have begun to shed some light on these questions, particularly in human islets. For example, antibody-based studies have identified four apparent subgroups of beta cells differing in the presence of cell-surface markers and in their transcriptome [11]. In parallel, optogenetic studies have identified a subpopulation of beta cells that coordinate islet-wide beta cell activity [12]. Recently, it has become feasible to interrogate the entire transcriptome of individual islet cells using massively parallel sequencing (next generation sequencing [NGS]). Each of these technologies has its own pitfalls, advantages and limitations, but taken together they provide an opportunity to better understand the differences and similarities among different islet cell types, and identify and study multiple subgroups of the major cell types. Furthermore, rare cell types that would be completely missed in bulk analysis can be readily captured and analysed using single-cell technologies. Here we provide a concise overview of these recent developments in the area of beta cell research and the implications for our understanding of beta cell biology and pathology.
Recent evidence for functional heterogeneity
Functional characteristics vary in islets and beta cell populations
Several recent studies have examined heterogeneity of beta cells under different conditions, using various technologies. Olsson and Carlsson [13] found that 20–25% of the islets in intact rat pancreases were hypoxic and that this percentage correlated with beta cell mass. Partial pancreatectomy decreased the percentage of hypoxic islets, whereas the fraction of hypoxic islets rose in the endogenous pancreas after beta cell mass was increased via pancreas transplantation. The authors further showed reduced metabolic activity in hypoxic islets, as measured by protein biosynthesis, suggesting that they represent a ‘dormant’ subpopulation that can be oxygenated and recruited to action upon metabolic demand. The functional significance of the hypoxic islet subpopulation is not clear and the stability of this population is not known: is hypoxia a long-standing feature of a subpopulation of islet cells or a highly dynamic state? In any case, this provocative suggestion of metabolic heterogeneity at the whole islet level certainly merits further investigation.
More recently, Hodson’s et al used in situ calcium imaging approaches, together with large scale map** of cellular connectivity, to reveal functional differences between beta cells in intact mouse and human islets. First, they identified a subpopulation of glucagon-like peptide-1 (GLP-1)-responsive beta cells that promoted coordinated beta cell activity [14]. GLP-1-induced beta cell interconnectivity was disrupted by NEFA exposure and negatively associated with donor BMI, suggesting that this heterogeneity might be relevant for type 2 diabetes pathogenesis. The same group went on to show that glucose stimulation of mouse and human islets led to the presence of a small number of hub cells (<10% of islet beta cells) with long-range connectivity resulting in downstream regulation of follower cells [12]. To investigate the functional role of these hubs, the authors used an optogenetic strategy to transiently silence individual hub or follower beta cell activity. Silencing hub beta cells (but not follower cells) disrupted islet functional connectivity, calcium dynamics and insulin secretion. The mechanisms responsible for this apparent intra-islet connectivity are not fully understood but may be mediated through gap junctions and other direct cell–cell communication pathways, as recently reviewed by Benninger and Piston [15]. Beta cell hubs were further shown to display higher mitochondrial membrane potential and features of less-mature beta cells when compared with follower cells. Finally, to mimic a diabetes setting, exposure to a cocktail of cytokines and gluco/lipotoxicity disrupted hub cell function, suggesting a link between hub cell dysfunction and beta cell failure in diabetes [12]. So far, no data exists on the transcriptome or proteome of hub cells, nor on whether all beta cells are endowed with the potential to become hub cells at some point. Potentially, single-cell transcriptome analysis (discussed below) may be used to identify a subpopulation of beta cells that carry the hallmarks of hub cells (e.g. high levels of glucokinase expression). Regardless, the idea that a small group of beta cells uniquely affects the behaviour of a larger population is highly provocative and may link beta cell heterogeneity with disease susceptibility for the first time.
Differences in gene expression in beta cell subpopulations
Bader et al [16] observed that adult mouse beta cells can be divided into two subpopulations based on their expression of a Wnt/planar cell polarity (PCP) downstream effector, known as flattop (FLTP). They generated an Fltp knock-in fluorescent reporter mouse model and used it to monitor Fltp expression in mouse islets throughout development. They found that the percentage of FLTP+ beta cells increases during postnatal development, reaching a maximum of 80% in adult mice. Interestingly, Fltp expression is also reduced in beta cells from humans with impaired glucose tolerance and type 2 diabetes compared with healthy individuals. Re-aggregated pseudo-islets originating from sorted murine FLTP+ cells secreted more insulin upon glucose stimulation than their FLTP− counterparts. In addition, the FLTP− fraction displayed higher proliferation rates, both in normal expansion of beta cells during early development and during pregnancy, a physiological instance of transient increased insulin demand. In line with this observation, we have shown that genes that are expressed more highly throughout beta cell development (including genes regulating beta cell function) are relatively downregulated in replicating beta cells [17].
While FLTP is not required for normal beta cell development, proliferation or function in mice, transcriptome analysis of FLTP+ and FLTP− mouse beta cell subtypes showed that FLTP expression is associated with a more mature beta cell state, suggesting that non-canonical Wnt signalling might be involved in beta cell maturation. Although larger, deeper sequencing datasets will be required to validate these initial findings, Bader et al suggest that, while FLTP does not regulate beta cell differentiation or maturation, it may represent a bona fide beta cell maturation marker.
Dorrell et al [11] developed antibodies against human islet cell-surface proteins and identified four subpopulations of beta cells based on the expression of two different cell-surface markers, CD9 and ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 1 (ST8SIA1). While most beta cells in healthy humans are CD9−/ST8SIA1−, the distribution of beta cell subtypes was altered in type 2 diabetes. This study suggests the exciting possibility that in diabetes there is a change in the balance between different beta cell types. As with other studies that suggest beta cell heterogeneity exists, it remains to be seen whether the four subpopulations identified by Dorrell et al reflect stable subtypes or dynamic states.
These recent studies identified major subgroups of beta cells based on specific functional characteristics (hypoxia or calcium signalling) or specific gene expression (Fltp, CD9 or ST8SIA1). While they all raise the possibility that functional heterogeneity exists among beta cells, more work will be required to understand how stable these populations are and to what extent they play a role in the pathogenesis of diabetes. The emerging technology of single-cell transcriptomics, discussed below, may provide a powerful tool to merge functional and molecular studies of beta cell heterogeneity.
Single-cell technologies for the study of human and rodent beta cell heterogeneity
To further explore beta cell heterogeneity, several groups have used cutting-edge single-cell technologies to identify rare, as well as common, beta cells subtypes.
Single-cell time-of-flight mass spectroscopy
Single-cell time-of-flight mass spectroscopy (CyTOF) is a rapidly evolving technology, with more protein-specific probes continuously being developed to provide more in-depth phenoty** of individual cells. Furthermore, the concept is being adapted for use on whole tissues, providing subcellular resolution and allowing for phenoty** of individual cells in situ. This methodology generates data analogous to multi-channel immunofluorescence but with the ability to simultaneously analyse a large number of antigens, far beyond what is possible with standard immunofluorescence approaches. Further, development of this technology may help to address questions related to the interaction between local environment (e.g. cell–cell interactions) and individual cell heterogeneity.
Wang et al used CyTOF to simultaneously analyse the expression of 24 proteins at single-cell resolution in human islet cells [18]. The authors subdivided beta cells from multiple donors into three main clusters, with proliferating Ki-67+ cells (which also had higher expression levels of other cell-proliferation markers), sitting in two of these clusters. Interestingly, one of the subtypes was poorly represented in the beta cells from type 2 diabetes donors, suggesting that this subtype may play a role in the pathogenesis of type 2 diabetes.
Massively parallel sequencing
Massively parallel sequencing is another technology undergoing rapid evolution, with ever-decreasing input requirements and plummeting costs. In the last 2 years, several groups have published initial findings evaluating the transcriptome of a large number of individual islet cells. Additional studies are expected in the coming years. There are a number of caveats that must be considered when designing and evaluating single-cell transcriptome experiments. First and foremost is the limited ability to accurately and consistently identify rare transcripts. All methods for massively parallel sequencing rely on an initial amplification step and none of the methods currently available can reproducibly amplify every single transcript in a cell. Assuming reasonable efficiency and linearity of amplification, the quantity of abundant transcripts can be reliably measured. Rare transcripts, on the other hand, may be randomly included or excluded in the initial amplification, resulting in over-estimation of the degree of heterogeneity between cells. This issue can be partially addressed by improving the precision of reverse transcription and the amplification step, and by computationally combining rare transcripts into functional groups so that the total number of transcripts in each group is sufficiently abundant to measure reliably.
Transcriptomic evidence for beta cell heterogeneity
When designing single-cell transcriptome experiments, there is an obvious trade-off between the number of cells examined, the depth of sequencing for each cell and the cost of the experiment. Both large numbers of cells and in-depth sequencing are required to identify and fully characterise rarer subgroups of beta cells. Li and colleagues [19] were the first to perform single-cell RNA sequencing on human islets. Their study, which interrogated only 70 cells, validated previously described marker genes at the single cell level and identified transcription factors that were specifically expressed in all islet cell types. While their findings provided early evidence for transcriptional heterogeneity, the number of cells interrogated was too small to thoroughly characterise subgroups within the different cell types.
Since Li et al’s study, several other groups have published single-cell transcriptomic data on an increasing numbers of islet cells from control and type 2 diabetes donors [20,21,22,23]. New and interesting data is emerging from these studies. For example, **n et al identified approximately 250 genes that were differentially expressed in type 2 diabetes, 92% of them not previously associated with islet cell function or growth [23]. Muraro et al [21] reported that the most significant genes dictating beta cell heterogeneity were SRXN1, SQSTM1, and three ferritin subunits, FTH1P3, FTH1, and FTL, which were all implicated in response to endoplasmic reticulum (ER) and oxidative stress [24, 25].
Recently, two groups sequenced large numbers of cells at relatively low sequence depth: Baron et al and Segerstolpe et al [20, 22]. Baron et al identified that variation in the expression of certain genes correlated with beta cell function (e.g. UCN3) and ER stress (HERPUD1, HSPA5 and DDIT3) [20]. Interestingly, this is in line with recent evidence showing that human and mouse beta cells exhibiting active ER stress as a result of high demand for insulin secretion are more likely to proliferate [26]. The authors further describe a substructure in the human ductal cell population; ductal cells are known to exhibit two morphologies, one forming the terminal duct and the other connecting to the acinus (centroacinar cells). Using immunostaining, the authors demonstrated spatial separation between cell types that corresponded to the transcriptome analysis. This study demonstrated the importance of correlating results provided by technologies that examine isolated cells with those that provide data on the local environment.
Segerstolpe and colleagues [22] reported subpopulations among alpha, beta and acinar cells. Among alpha-cells, they detected 12 proliferating cells belonging to multiple donors (both healthy and with type 2 diabetes) that expressed high levels of proliferation-associated genes (e.g. TOP2A, MKI67, CENPF, BIRC5 and CDK1), without a change in glucagon expression. The gene expression signatures of these rare proliferating cells may provide clues to the mechanisms of self-renewal of human pancreatic tissue. Segerstolpe and colleagues also described five beta-cell sub-clusters with combinatorial expression of RBP4, GPR120 (also known as FFAR4), ID1, ID2 and ID3. Although transcriptionally different, cells in the five clusters expressed INS at similar levels [22]. An increase in circulating levels of RBP4 is a prominent feature of obesity and type 2 diabetes and this correlates with insulin resistance in the periphery [27]. The RBP4-expressing beta cell clusters also expressed GPR120, which is shown to induce insulin release in mouse islets [28]. The remaining clusters were characterised by expression of ID1 and ID3, regulators of basic helix-loop-helix (bHLH) transcription factors [29]. Segerstolpe et al also identified two clusters of acinar cells, one characterised by elevated expression of inflammation-related genes and the other expressing higher levels of key acinar genes, encoding secretory digestive enzymes and their transcription factor regulators [22].
Our research group [30] has also generated transcriptomes of human islet cells from a range of organ donors, including children, adults, and type 1 and type 2 diabetes donors, sequencing each cell at much greater depth compared with other studies. We discovered that a substantial number of islet cells display divergent expression profiles within each canonical endocrine cell type. For instance, we found that insulin mRNA levels in beta cells can vary by more than two orders of magnitude from cell to cell. We further described the expression profile of a rare proliferating alpha cell on the basis of Ki67 (also known as MKI67) expression. Many key hedgehog signalling genes were activated in this replicating cell, in addition to the expected gene signatures of cell cycle regulation and checkpoint control.
The studies described here demonstrate the power of emerging single-cell RNA sequencing technology for evaluating the interplay of different physiological programmes (for example proliferation and function) at the single-cell level and changes in relative abundance of different cell subtypes in various pathologies. Transcriptome data will have to be combined with functional studies to determine the functional significance of transcriptome variability.
One of the open questions in the field of beta cell plasticity is the frequency and significance of trans-differentiation between different canonical cell types. In an attempt to address this issue, single-cell transcriptomic studies have encountered an important technical problem. While several methods are available to capture individual cells for sequencing, none are 100% reliable. Therefore, all of the studies described above used methods to exclude data that could have been generated from more than one cell. For example, a transciptome that includes similar levels of glucagon and insulin gene could represent a cell that is in transition between the alpha- and beta cell phenotypes or a doublet of different cells inadvertently captured in the same assay. Applying too stringent criteria to exclude potential doublets will beg the question of the presence of bihormonal cells, since these cells, if they exist, will be excluded from analysis. Additional work, applying existing and novel approaches to reliably identify true doublets, will be required to address the issue of multihormonal cells.
Multihormonal islet cells
For many years, biologists have had at their disposal a powerful method to interrogate beta cells in their niche and the genes that they express at the single-cell level. Combined with immunostaining, histological analysis can provide such information, with the caveats that confocal microscopy is required to prevent an incidental overlap of two different cells in the focal plane, and that the number of proteins amenable for analysis in the same cell is usually limited to about four (the number of laser channels in a typical confocal microscope). One particular form of beta cell heterogeneity observed in co-immunostaining experiments is the co-expression of insulin and other hormones in a small fraction of beta cells. Double-hormone positive cells are frequent in early fetal pancreas development, but are thought to represent a ‘dead end’ for the cell, with it dying before birth [31]. During postnatal life, such cells are rarely observed, and their biological significance is not clear. The incidence of double-hormone positive cells was proposed to increase in type 2 diabetes, and to represent a new mode of beta cell failure in which beta cells lose their identity or dedifferentiate [32]. Consistent with this suggestion, we have recently reported that, in type 2 diabetes, a subset of pre-existing beta or delta cells induce the expression of the fetal islet hormone gastrin [33]. In addition, deletion of key transcription factors in adult beta cells leads to the appearance of beta cells co-expressing insulin and other hormones, or even converting entirely to other cell types [34,35,36]. In the reverse direction, Herrera and colleagues have shown that near-total ablation of beta cells leads to the reprogramming of a subset of alpha cells or delta cells to acquire beta cell identity [37, 38]. All these studies suggest a new plastic view of islet cells, in which different islet cell types are closely related to each other and can inter-convert under certain conditions. While beta cell identity transitions are being extensively studied, virtually nothing is known about why only a small subset of islet cells demonstrates plasticity. Is plasticity an inherent trait of a minority population of islet cells, or are all islet cells amenable to identity changes given the right stimulus or the right micro-environment? Answers to these questions may link beta cell heterogeneity to tissue dynamics and greatly impact on research into beta cell regeneration. As with the other observations of beta cell heterogeneity, one key question with regards to multihormonal islet cells is whether differences in phenotype represent cell types or cell states.
Notably, recent technological advances are merging omics with histological analysis; these approaches are known as ‘spatial genomics’ [39] or ‘spatial proteomics’ [40, 41]. Single-cell RNA sequencing or mass spectrometry in situ (preserving the spatial location of the studied cells) promises to expose new layers of beta cell biology and heterogeneity. Furthermore, technologies are emerging that capture epigenomic features, such as DNA methylation, chromatin accessibility and histone modifications at the single cell level, adding additional dimensions to the study of cellular plasticity and diversity.
Summary and conclusions
The studies described above are the first to use the emerging technologies of single-cell transcriptomics and proteomics to study pancreatic islet cells. They demonstrate the feasibility of implementing these technologies in the study of beta cells and give a glimpse of the type of data that will become available in the coming years. Obtaining detailed information from a large number of cells from many individuals with different pathologies will permit the in-depth study of very rare cell types, such as actively replicating beta cells, including their division programme, the stages of this process, vulnerabilities (e.g. activation of stress response pathways) and potential points for intervention, with the aim of increasing functional beta cell mass. A better understanding of beta cell heterogeneity may also reveal a disproportionally large contribution of a small subset of cells to overall beta cell function and to beta cell failure in diabetes.
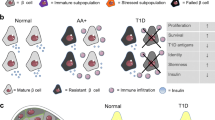
There are still many open questions (Fig. 1); for example, do the different human beta cell subtypes reflect distinct developmental stages and/or stable subpopulations rather than inter-changeable cellular states? Clever study designs are needed to answer this question since classical lineage tracing studies are obviously not possible in humans. Using live-cell imaging, Szabat et al provide evidence that beta cells can change transcriptional profiles over time, suggesting the possibility of at least some plasticity in respect to subtype identity [42]. The theoretical significance of this is great, since manipulations aimed at switching from one subtype to another may prove more feasible than current efforts aimed at increasing beta cell number.

Schematic representation of open questions related to the origin, status and stress-mediated fate of four hypothetical beta cell subtypes, each represented by a different colour. (a) The origin of beta cell heterogeneity. Are the different beta cell subtypes stable or are they able to switch from one phenotype to another? If so, what signals regulate such switches: metabolic demand, pregnancy, age? (b) Distribution of beta cell subtypes. Do different beta cell subtypes interact within islets? Are they clustered in different islets (perhaps in different regions of the pancreas) or are they scattered randomly among the islets? (c) Effects of metabolic stress on beta cell heterogeneity. When stressed, how do different beta cell subtypes react? Do they undergo apoptosis, senescence, subtype plasticity or trans-differentiation to non-beta cells (the last is not shown)? Are some subtypes more sensitive or resistant to specific stresses?
We may also ask: do different subtypes randomly scattered throughout the pancreas or are they located in specific locations within the pancreas or islets? By integrating in-depth transcriptome and proteome data from dispersed cells with spatial proteomic technologies, such as imaging mass cytometry, detailed map** of different cell types will be possible. Further, do the specific subtypes differ in their vulnerability to the metabolic stress that is inherent to type 2 diabetes? In animal models, these questions can be answered by sophisticated lineage tracing experiments. In humans obtaining answers will be more challenging. Also, does altered distribution of beta cell subtypes in type 2 diabetes contribute to beta cell dysfunction and can our understanding of the different subgroups identify novel therapeutic targets for the treatment or prevention of diabetes?
These critical questions may be answered, at least in part, by integrating data from multiple groups, generated using different technologies in different sample sets, to obtain a comprehensive picture of islet-cell response to physiological and pathological states. It is eminently clear that, with these emerging technologies, we are at the brink of a new era in our understanding of islet cell function. Time will tell whether this new information can be translated into clinically relevant medical advances.
Abbreviations
- CyTOF:
-
Time-of-flight mass spectroscopy
- ER:
-
Endoplasmic reticulum
- FLTP:
-
Flattop
- GLP-1:
-
Glucagon-like peptide-1
- ST8SIA1:
-
ST8 alpha-N-acetyl-neuraminide alpha-2,8-sialyltransferase 1
References
Roscioni SS, Migliorini A, Gegg M, Lickert H (2016) Impact of islet architecture on β-cell heterogeneity, plasticity and function. Nat Rev Endocrinol 12:695–709
Hellerstrom C, Petersson B, Hellman B (1960) Some properties of the B cells in the islet of Langerhans studied with regard to the position of the cells. Acta Endocrinol 34:449–456
Salomon D, Meda P (1986) Heterogeneity and contact-dependent regulation of hormone secretion by individual B cells. Exp Cell Res 162:507–520
Bosco D, Meda P (1991) Actively synthesizing beta-cells secrete preferentially after glucose stimulation. Endocrinology 129:3157–3166
Pipeleers D, Kiekens R, Ling Z, Wilikens A, Schuit F (1994) Physiologic relevance of heterogeneity in the pancreatic beta-cell population. Diabetologia 37:S57–S64
Van Schravendijk CF, Kiekens R, Pipeleers DG (1992) Pancreatic beta cell heterogeneity in glucose-induced insulin secretion. J Biol Chem 267:21344–21348
Pipeleers DG (1992) Heterogeneity in pancreatic beta-cell population. Diabetes 41:777–781
Elayat AA, el-Naggar MM, Tahir M (1995) An immunocytochemical and morphometric study of the rat pancreatic islets. J Anat 186:629–637
Wang X, Misawa R, Zielinski MC et al (2013) Regional differences in islet distribution in the human pancreas—preferential beta-cell loss in the head region in patients with type 2 diabetes. PLoS One 8:e67454
Poudel A, Savari O, Striegel DA et al (2015) Beta-cell destruction and preservation in childhood and adult onset type 1 diabetes. Endocrine 49:693–702
Dorrell C, Schug J, Canaday PS et al (2016) Human islets contain four distinct subtypes of β cells. Nat Commun 7:11756
Johnston NR, Mitchell RK, Haythorne E et al (2016) Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metab 24:389–401
Olsson R, Carlsson P-O (2011) A low-oxygenated subpopulation of pancreatic islets constitutes a functional reserve of endocrine cells. Diabetes 60:2068–2075
Hodson DJ, Mitchell RK, Bellomo EA et al (2013) Lipotoxicity disrupts incretin-regulated human β cell connectivity. J Clin Invest 123:4182–4194
Benninger RKP, Piston DW (2014) Cellular communication and heterogeneity in pancreatic islet insulin secretion dynamics. Trends Endocrinol Metab 25:399–406
Bader E, Migliorini A, Gegg M et al (2016) Identification of proliferative and mature β-cells in the islets of Langerhans. Nature 535:430–434
Klochendler A, Caspi I, Corem N et al (2016) The genetic program of pancreatic beta-cell replication in vivo. Diabetes 65:2081–2093
Wang YJ, Golson ML, Schug J et al (2016) Single-cell mass cytometry analysis of the human endocrine pancreas. Cell Metab 24:616–626
Li J, Klughammer J, Farlik M et al (2016) Single-cell transcriptomes reveal characteristic features of human pancreatic islet cell types. EMBO Rep 17:178–187
Baron M, Veres A, Wolock SL, et al. (2016) A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst 3:346–360
Muraro MJ, Dharmadhikari G, Grün D et al (2016) A single-cell transcriptome atlas of the human pancreas. Cell Syst 3:385–394
Segerstolpe Å, Palasantza A, Eliasson P (2016) Single-cell transcriptome profiling of human pancreatic islets in health and type 2 diabetes. Cell Metab 24:593–607
**n Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C (2016) RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab 26:608–615
Orino K, Lehman L, Tsuji Y, Ayaki H, Torti SV, Torti FM (2001) Ferritin and the response to oxidative stress. Biochem J 357:241–247
Zhou Y, Duan S, Zhou Y et al (2015) Sulfiredoxin-1 attenuates oxidative stress via Nrf2/ARE pathway and 2-Cys Prdxs after oxygen-glucose deprivation in astrocytes. J Mol Neurosci 55:941–950
Sharma RB, O'Donnell AC, Stamateris RE et al (2015) Insulin demand regulates β cell number via the unfolded protein response. J Clin Invest 125:3831–3846
Yang Q, Graham TE, Mody N et al (2005) Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 436:356–362
Moran BM, Abdel-Wahab YHA, Flatt PR, McKillop AM (2014) Evaluation of the insulin-releasing and glucose-lowering effects of GPR120 activation in pancreatic β-cells. Diabetes Obes Metab 16:1128–1139
Ling F, Kang B, Sun X-H (2014) Chapter five - Id proteins: small molecules, mighty regulators. In: Reshma T (ed) Current topics in developmental biology. Academic Press, Waltham, MA, pp 189–216
Wang YJ, Schug J, Won K-J et al (2016) Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65:3028–3038
Herrera PL (2000) Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 127:2317–2322
Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 150:1223–1234
Dahan T, Ziv O, Horwitz E et al (2017) Pancreatic beta-cells express the fetal islet hormone gastrin in rodent and human diabetes. Diabetes 66:426–436
Ediger BN, Lim H-W, Juliana C et al (2016) LIM domain–binding 1 maintains the terminally differentiated state of pancreatic β cells. J Clin Invest 127:215–229
Gutiérrez GD, Bender AS, Cirulli V et al (2016) Pancreatic β cell identity requires continual repression of non–β cell programs. J Clin Invest 127:244–259
Swisa A, Avrahami D, Eden N et al (2016) PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J Clin Invest 127:230–243
Chera S, Baronnier D, Ghila L et al (2014) Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 514:503–507
Thorel F, Népote V, Avril I et al (2010) Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 464:1149–1154
Stahl PL, Salmen F, Vickovic S et al (2016) Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 353:78–82
Rizzo DG, Prentice BM, Moore JL, Norris JL, Caprioli RM (2017) Enhanced spatially resolved proteomics using on-tissue hydrogel-mediated protein digestion. Anal Chem 89:2948–2955
Spraggins JM, Rizzo DG, Moore JL, Noto MJ, Skaar EP, Caprioli RM (2016) Next-generation technologies for spatial proteomics: integrating ultra-high speed MALDI-TOF and high mass resolution MALDI FTICR imaging mass spectrometry for protein analysis. Proteomics 16:1678–1689
Szabat M, Pourghaderi P, Soukhatcheva G et al (2014) Kinetics and genomic profiling of adult human and mouse β-cell maturation. Islets 3:175–187
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Related work in our laboratories is supported through NIH grants (UC4DK104119; to BG and DA), the BIRAX Regenerative Medicine Initiative (14BX14NHBG; to BG) and the Israel Science Foundation–JDRF Joint Program in Type 1 Diabetes Research (1506/12; to BG).
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
All authors were responsible for drafting the article and revising it critically for important intellectual content. All authors approved the version to be published.
Electronic supplementary material
ESM Downloadable slide
(PPTX 272 kb)
Rights and permissions
About this article

Cite this article
Avrahami, D., Klochendler, A., Dor, Y. et al. Beta cell heterogeneity: an evolving concept. Diabetologia 60, 1363–1369 (2017). https://doi.org/10.1007/s00125-017-4326-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-017-4326-z