
Abstract
Aims/hypothesis
HNF1A-MODY is a monogenic form of diabetes caused by mutations in the HNF1A gene. Here we identify, for the first time, HNF1A-MODY-associated microRNAs (miRNAs) that can be detected in the serum of HNF1A-MODY carriers.
Methods
An miRNA array was carried out in rat INS-1 insulinoma cells inducibly expressing the common human Pro291fsinsC-HNF1A frame shift mutation. Differentially expressed miRNAs were validated by quantitative real-time PCR. Expression of miRNAs in the serum of HNF1A-MODY carriers (n = 31), MODY-negative family members (n = 10) and individuals with type 2 diabetes mellitus (n = 17) was quantified by absolute real-time PCR analysis.
Results
Inducible expression of Pro291fsinsC-HNF1A in INS-1 cells caused a significant upregulation of three miRNAs (miR-103, miR-224, miR-292-3p). The differential expression of two miRNAs (miR-103 and miR-224) was validated in vitro. Strongly elevated levels of miR-103 and miR-224 could be detected in the serum of HNF1A-MODY carriers compared with MODY-negative family controls. Serum levels of miR-103 distinguished HNF1A-MODY carriers from HbA1c-matched individuals with type 2 diabetes mellitus.
Conclusions/interpretation
Our study demonstrates that the pathophysiology of HNF1A-MODY is associated with the overexpression of miR-103 and miR-224. Furthermore, our study demonstrates that these miRNAs can be readily detected in the serum of HNF1A-MODY carriers.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
HNF1A-MODY is a monogenic form of diabetes mellitus [1] caused by mutations in the gene encoding transcription factor 1 (TCF-1)/hepatocyte nuclear factor 1α (HNF1A), the most common of which is a frame shift mutation in exon 4, Pro291fsinsC-HNF1A [2]. Aberrant gene regulation is a key feature in HNF1A-MODY [3–5]. Previous reports on gene expression profiling performed in in vitro and in vivo models of HNF1A-MODY have identified defects in insulin secretion, glucose metabolism, mitochondrial electron transport, intercellular communication and cell adhesion, apoptosis and proliferation to be associated with HNF1A-MODY [6–11]. MicroRNAs (miRNAs) have emerged as potent regulators of gene function in numerous diseases including cancer and diabetes [12–15]. MicroRNAs are typically 19–25 nucleotide non-coding RNA molecules, which suppress gene expression via imperfect base pairing to the 3′ untranslated region (3′UTR) of target mRNAs, leading to repression of protein production or mRNA degradation [16, 17]. So far, more than 1,500 predicted miRNA precursors, leading up to 2,154 mature miRNAs, have been identified in the human genome (www.miRbase.org; accessed November 2012). These findings suggest that miRNAs play a fundamental role in genome-wide regulation of gene expression.
Recent studies provided evidence that miRNAs have an important role in pancreatic development [18] and beta cell differentiation [19], as well as in insulin production, secretion and action [15]. To date there have not been any studies investigating the potential involvement of miRNAs in the pathogenesis of HNF1A-MODY. In this study we therefore investigated changes in miRNA expression in INS-1 insulinoma cells inducibly expressing the Pro291fsinsC-HNF1A hot-spot mutation and in serum from individuals with HNF1A-MODY.
Methods
INS-1 cells overexpressing HNF1A in an inducible system
Rat INS-1 insulinoma cells inducibly expressing the human HNF1A-MODY frame shift mutant Pro291fsinsC-HNF1A under the control of a doxycycline-dependent transcriptional activator have been described previously [3, 9, 10, 20]. The Pro291fsinsC-HNF1A mutant has been shown to bind endogenous HNF1A, and to act as a dominant-negative transcription factor in vitro [4]. Cells were cultured in RPMI 1640 at 6 mmol/l glucose supplemented with 10% FBS (PAA, Cölbe, Germany), 2 mmol/l l-glutamine, 1 mmol/l pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mmol/l HEPES (pH 7.4) and 50 μmol/l 2-mercaptoethanol (Sigma, Dublin, Ireland) [21]. Measurement of HNF1A induction in INS-1 cells was carried out using absolute quantitative PCR. An HNF1A gene-specific PCR amplicon was prepared as a standard. The calibration curve was created by plotting the threshold cycle (Ct) corresponding to each standard vs their corresponding log number of HNF1A standard (expressed as cDNA copy number of the HNF1A gene). Normalisation was carried out using total RNA values, as previously described [9].
TaqMan microRNA array
INS-1 cells were cultured in T25 flasks in complete medium with or without 500 ng/ml doxycycline for 24 h or 48 h. Cells were harvested for total RNA using the Qiagen miRNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. All downstream applications were performed on ice in an RNAse-free environment to prevent miRNA degradation. Thirty nanograms of total RNA enriched in miRNAs was converted to cDNA using the TaqMan MicroRNA Reverse Transcription Kit (Roche-Applied Biosystems, Burgess Hill, UK), and pre-amplification was carried out on the cDNA using Megaplex PreAmp Primers (Invitrogen, Paisley, UK). TaqMan Universal PCR Master Mix (Roche-Applied Biosystems) was added to the samples, which were then loaded onto TaqMan Rodent MicroRNA A+B Cards Set v2.0 (Invitrogen) and run on a 7900HT system using 7500 SDS version 1.2.2 (Life Technologies, Carlsbad, CA, USA). Each card contained three endogenous rat controls. The data was normalised to the endogenous control MammU6 (MammU6-4395470). The controls U87 (U87-4386735) and Y1 (Y1-4386739) were normalised to the MammU6. Before calculating relative expression values, normalisation was carried out by subtracting the average endogenous control sample Ct from the individual miRNA Ct values. Relative expression (RQ) of miRNA was calculated using the ∆∆Ct method; \( \mathrm{RQ}={2}^{-\varDelta \varDelta {\mathrm{C}}_{\mathrm{t}}} \). p values were calculated using a t test assuming two-tailed distribution in two samples of unequal variance.
Real-time quantitative PCR analysis
INS-1 cells were cultured in six-well plates in complete medium with or without 500 ng/ml doxycycline for 24 h or 48 h. Cells were harvested for RNA enriched in miRNAs using the Qiagen miRNeasy kit according to the manufacturer’s protocol. Reverse transcription was carried out on 30 ng of RNA using the TaqMan MicroRNA Reverse Transcription Kit and Taqman probes specific to the individual miRNAs chosen (miR-103, miR-224 and miR-29a). Real-time PCR amplification was performed with the reverse transcription products using TaqMan 2× Universal PCR Master Mix without UNG Amperase, and miRNA-specific TaqMan probes were purchased from Roche Diagnostics, Burgess Hill, UK: hsa-miR-103 (AGCAGCAUUGUACAGGGCUAUGA); hsa-miR-224 (CAAGUCACUAGUGGUUCCGUU); hsa-miR-29a (UAGCACCAUCUGAAAUCGGUUA).
As expression of miRNA-29a did not show any change in the miRNA array it was chosen as an internal control for the real-time PCR in INS-1 cells overexpressing the Pro291fsinC-HNF1A mutant. Samples were run on an Applied Biosystems 7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA) with an initial denaturation at 95°C for 10 min, followed by 40 cycles at 95°C for 15 s and 60°C for 1 min.
Prediction of gene targets
Potential gene targets of differentially expressed miRNAs were predicted using two public databases: miRanda (www.microrna.org/microrna/home.do) accessed April 2010 and TargetScan (www.targetscan.org/), accessed April 2010. The gene functional classification tool DAVID (http://david.abcc.ncifcrf.gov/home.jsp, accessed June 2010) was used to identify the signalling pathways to which the predicted gene targets belonged [22, 23]
Participants and clinical and laboratory measurements
Individuals with a clinical diagnosis of MODY were recruited from the MODY diabetes clinics in the Mater Misericordiae University Hospital Dublin in Ireland. Sequencing of the HNF1A gene was performed by IntegraGen (Bonn, Germany) in 2006–2007 and the Molecular Genetics Laboratory (Exeter, UK) in 2008–2010. Genetically confirmed MODY carriers included 31 cases with HNF1A mutations. The individuals with HNF1A mutations were from 11 pedigrees and the mutations included L17H (n = 1), G207D (n = 1), P291finsC (n = 14), S352fsdelG (n = 9), F426X (n = 2), P379T (n = 2) and IVS7-6G>A (n = 2). The mutations named above have been previously published and are known to co-segregate in families with diabetes [24, 25]. There were ten normoglycaemic BMI-matched HNF1A-MODY-negative family members available as a control group. In addition, 17 participants with type 2 diabetes mellitus, matched for HbA1c, were recruited from the diabetes clinics in the Mater Misericordiae University Hospital.
All participants underwent a clinical assessment, including a full medical history and physical examination. Details of the participants’ weight, height and blood pressure were recorded. A 75 g OGTT was performed on participants after a 12 h overnight fast, with measurement of glucose, insulin and C-peptide at baseline and at 30 min intervals for 120 min to determine the degree of glucose tolerance and insulin secretory response. In patients with diabetes, oral hypoglycaemic agents were stopped at least 48 h before the OGTT while in those taking insulin, long-acting insulin therapy was stopped for 24 h and short-acting insulin stopped for 12 h before OGTT. ADA diagnostic criteria were used to define the degree of glucose tolerance. The oral glucose insulin sensitivity (OGIS) was calculated as previously described [26]. The plasma glucose concentration was measured using Beckman Synchron DXC800 (Beckman Instruments, Brea, CA, USA). HbA1c was determined using high-performance liquid chromatography (HA81-10; Menarini, Rome, Italy). Insulin and C-peptide were analysed using Immulite 2000 immunoassay (Siemens Healthcare Diagnostics, Deerfield, IL, USA).
The study was approved by the Research Ethics Committee at the Mater Misericordiae University Hospital Dublin and all participants gave informed written consent.
RNA isolation from serum and absolute quantitative RT-PCR assays
Total RNA enriched with miRNAs was isolated from serum samples (200 μl), taken from HNF1A-MODY carriers, participants with type 2 diabetes mellitus and MODY-negative normoglycaemic family members, using the Total RNA Purification Kit (Norgen Biotek, Thorold, ON, Canada). Reverse transcription was carried out on 10 ng of RNA using the TaqMan MicroRNA Reverse Transcription Kit and Taqman probes specific to the individual miRNAs (miR-103 and miR-224). Individual miRNAs from participants’ serum samples were detected by absolute qPCR. TaqMan assays for human miRNAs hsa-miR-103 and hsa-miR-224 were obtained from Applied Biosystems. Oligoribonucleotides corresponding to the mature sequence of each miRNA were synthesised (Sigma Aldrich, Wicklow, Ireland) and reverse transcribed to generate a standard curve. The oligoribonucleotides sequences used were: miR-103 mature sequence: AGCAGCAUUGUACAGGGCUAUGA; and miR-224 mature sequence: CAAGUCACUAGUGGUUCCGUU. The standard curve was established using cDNA generated from the 23-bp or 21-bp human miR-103/miR-224 mature sequence amplicons, respectively. Two microlitres of cDNA were used as templates for PCR using the LightCycler 2.0 (Roche-Applied Biosystems). The miR-103 and miR-224 amplicons were isolated and purified using QIAquick PCR purification kit (Qiagen). The absolute quantity of the purified amplicons was measured by their absorbance at 260 nm and converted to the number of copies using the molecular weight of DNA. The miR-103 and miR-224 amplicons were diluted to a concentration of 5.98 × 107/5.87 × 107 copies per μl (stock solution). When a standard curve was to be established for the real-time quantitative PCR assay, the stock solution was diluted over seven orders of magnitude (e.g. 5.98 × 107, 5.98 × 106, 5.98 × 105, 5.98 × 104, 5.98 × 103, 5.98 × 10 and 5.98 × 101, copies per μl). All unknown sample concentrations had to fall within this range. Samples were run on the LightCycler 2.0 system with an initial denaturation at 95°C for 10 min, followed by 80 cycles at 95°C for 10 s (denaturation), 60°C for 20 s (annealing) and 72°C for 1 s. The data were analysed using LightCycler Software 4.0. The normalised values (dCt) from serum of HNF1A-MODY carriers were compared with serum from MODY-negative, normoglycaemic family members and participants with type 2 diabetes mellitus.
Statistical analysis
Results of miRNA expression in cells were expressed as means±SEM. Differences between treatments were analysed by Student’s t test, as well as one-way ANOVA and subsequent Tukey’s tests. Serum measurements and clinical data were given as median and inter quartile range (IQR) and compared by Mann–Whitney U test and Spearman correlation analysis. Statistical analysis was conducted using SPSS version 19 (IBM, Armonk, NY, USA) and MatLab version 7.1 (MathWorks, Natick, MA, USA). Differences were considered to be significant at p < 0.05.
Results
MicroRNA expression profiling in INS-1 cells identifies specific miRNAs regulated by the MODY-associated Pro291fsinsC HNF1A hot-spot mutation
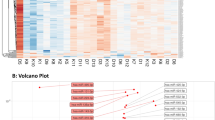
Expression of the dominant-negative HNF1A frame shift mutation Pro291fsinsC was induced in INS-1 cells by treatment with doxycycline for 24 h and 48 h to suppress endogenous HNF1A function. We observed a time-dependent induction of Pro291fsinsC-HNF1A mRNA (Fig. 1a). To identify specific miRNAs regulated by Pro291fsinsC-HNF1A in an unbiased manner, a TaqMan rodent miRNA array analysis was carried out. A total of 303 unique rat miRNAs were tested. Using a fold change cut-off of ≥1.5, we found three miRNAs (miR-103, miR-224, and miR-292-3p) to be differentially expressed at both time points, using biological duplicates, compared with an internal control (Fig. 1b). Analysis of the miRBase database (www.mirbase.org/index.shtml) [27] revealed that miR-292-3p is not expressed in humans, and was therefore not further investigated. We next validated the two upregulated miRNAs (miR-103 and miR-224) by individual TaqMan qRT-PCR assays. INS-1 cells were treated with doxycycline for 24 h and 48 h to induce overexpression of the Pro291fsinsC-HNF1A mutant [9]. Consistent with our profiling results, we observed that both miR-103 and miR-224 were significantly upregulated after 24 h and 48 h of exposure to doxycycline (Fig. 1c).

miR-103 and miR-224 are differentially expressed in INS-1 cells inducibly expressing Pro291fsinsC-HNF1A. (a) Time course of Pro291fsinC-HNF1A mRNA induction in INS-1 cells. Please refer to Methods section. (b) Identification of miRNA regulated by Pro291fsinC-HNF1A using rodent miRNA array plates. Data is represented as the RQ of two independent biological samples treated with doxycycline at 24 h (grey bars) and 48 h (black bars) time points relative to an internal control. (c) Validation of miRNA fold expression changes. ΔCt values were calculated by subtracting the Ct value for miR-29a (control) from the corresponding Ct value for each miRNA. White bars, control; grey bars, 24 h timepoint; black bars, 48 h time point. Experiments were carried out in triplicate (*p < 0.05 vs control)
Database analysis was carried out to determine possible gene targets of the experimentally validated, differentially expressed miRNAs. We employed the use of two different target prediction algorithms, miRanda and TargetScan, for our analysis. Gene functional analysis was carried out using DAVID to group putative gene targets into functional signalling KEGG pathways. The number of genes (count) and the percentage of genes from the predicted lists are shown in electronic supplementary material (ESM) Table 1. Analysis of putative target genes for miR-103 revealed the insulin signalling pathway to be among the top signalling pathways targeted, as well as Ca2+ signalling; analysis of putative target genes for miR-224 revealed endocytosis and TGF-β signalling.
MicroRNA-103 and miR-224 are detectable in the serum of HNF1A-MODY carriers
As miRNAs have been shown to be secreted from cells, we next investigated whether miR-103 or miR-224 levels were also elevated in the serum of HNF1A-MODY carriers compared with family controls. We performed an absolute quantitative PCR analysis of these two miRNAs extracted from individual serum from HNF1A-MODY patients (n = 31) and age- and BMI-matched MODY-negative family members (n = 10). Clinical characteristics are shown in Table 1.
We found miR-103 to be potently upregulated in the sera of the majority of HNF1A-MODY patients compared with the MODY-negative controls (Fig. 2; median [IQR] 1,030 [458–3,318] copy number vs 14 [0–126] copy number, p = 0.00001). We next explored whether levels of miR-103 correlated with any clinical or biochemical variables. Interestingly, miR-103 correlated negatively with triaclyglycerol and systolic blood pressure (ρ = −0.44, p = 0.02 and ρ = −0.45, p = 0.01), but did not correlate with HbA1c (ρ = 0.12, p = 0.54), OGIS (ρ = 0.18, p = 0.34), age (ρ = −0.21, p = 0.25) or duration of diabetes (ρ = −0.02, p = 0.92) (Table 2). Almost one-third (32.2%) of patients were treated with diet alone and the remainder were either treated with insulin or oral hypoglycaemic agents, predominantly sulfonylureas. The particular type of treatment did not correlate with levels of miR-103. The type of the HNF1A mutation (transactivation domain vs dimerisation domain) did not have a significant influence on miR-103 levels (transactivation domain vs dimerisation domain: median [IQR] 1,060 [913–1,848] copy number vs 660 [331–7,005] copy number, p = 0.5531).

HNF1A-MODY carriers show elevated serum levels of miR-103 and miR-224. miR-103 (black bars) is significantly higher in HNF1A-MODY than in MODY-negative family controls (median [IQR] 1,030 [458–3,318] copy number vs 14 [0–126] copy number, p = 0.00001). Serum levels of miR-224 (grey bars) are also elevated in HNF1A-MODY compared with family controls (median [IQR] 2,610 [9–18,475] copy number vs 33 [23–46] copy number, p = 0.0467)
Interestingly, miR-224 expression was also very abundant in the sera of HNF1A-MODY patients compared with the amount detected in the control group (Fig. 2; median [IQR] 2,610 [9–18,475] copy number vs 33 [23–46] copy number, p = 0.0467). In contrast to miR-103, levels of miR-224 correlated significantly with OGIS (ρ = 0.44, p = 0.0167) but did not correlate with any other clinical variables (TG ρ = −0.09, p = 0.64; systolic blood pressure [SBP] ρ = −0.02, p = 0.93; HbA1c ρ = −0.16, p = 0.39; age ρ = 0.09, p = 0.64; duration of diabetes ρ = −0.17, p = 0.35) (Table 2). The type of treatment used for glycaemic control did not correlate with the expression of miR-224. However, there was a significant correlation with the type of mutation (transactivation domain vs dimerisation domain: median [IQR] 16,100 [27–27,050] copy number vs 181 [3–9,800] copy number, p = 0.046). We also noted that HNF1A-MODY carriers with high levels of circulating miR-103 frequently showed low levels of miR-224, and vice versa (Fig. 2). Assuming that it may be sufficient that elevation of one of the miRNAs may be an important event in the pathophysiology of HNF1A-MODY, we also explored whether the sum of miR-224+miR-103 correlated with a clinical feature. Interestingly, the miR-224+miR-103 sum correlated positively with OGIS (ρ = 0.58, p = 0.001) and negatively with fasting glucose (ρ = −0.51, p = 0.0034).
HNF1A-MODY carriers have higher miR-103 serum levels than HbA 1c -matched individuals with type 2 diabetes mellitus
We next aimed to provide the first clinical proof-of-concept that the HNF1A-MODY-associated miRNAs identified may also distinguish HNF1A-MODY from other forms of diabetes. We therefore determined serum levels of miR-103, which showed the highest discriminative power in the above studies, in a cohort of HbA1c-matched individuals with type 2 diabetes mellitus. HNF1A-MODY carriers had a lower BMI than the individuals with type 2 diabetes mellitus (median [IQR] 24.4 [22.1–26.2] kg/m2 vs 26.8 [25.3–27.4] kg/m2, p = 0.04). HNF1A-MODY carriers had a better metabolic profile than the individuals with type 2 diabetes mellitus, with lower blood pressure recordings (SBP, median [IQR] 120.5 [114.0–130.0] mmHg vs 130.0 [127.5–143.8] mmHg, p = 0.02 and diastolic blood pressure [DBP] median [IQR] 70.5 [67.0–80.0] mmHg vs 80.0 [75.3–85.0] mmHg, p = 0.009) and higher HDL levels (median [IQR] 1.4 [1.1–1.7] mmol/l vs 1.1 [0.9–1.3 mmol/l], p = 0.03). Of note, miR-103 serum levels were also significantly higher in the sera of HNF1A-MODY carriers than in sera from the group with type 2 diabetes mellitus (Fig. 3; median [IQR] 1,030 [458–3,318] copy number vs 129 [8–399] copy number, p = 0.00003).

miR-103 may distinguish HNF1A-MODY carriers from individuals with type 2 diabetes mellitus. Box plot showing median and IQR; dashed line indicates mean of miRNA copy numbers. Serum levels of miR-103 are significantly lower in individuals with type 2 diabetes mellitus than in HNF1A-MODY (median [IQR]: circles, 129 [8–399] copy number vs triangles 1,030 [458–3,318] copy number, *p = 0.00003). Data are from the HNF1A-MODY carriers (n = 31) and a group of HbA1c-matched participants with type 2 diabetes mellitus (n = 17)
Discussion
Monogenic forms of diabetes, such as HNF1A-MODY, account for a small proportion of cases but their penetrance and pathogenesis over a 20 year timespan makes them attractive models to study disease progression and therapeutic responses, and to identify potential predictive or prognostic biomarkers. In this study, we characterised miRNA expression in INS-1 cells inducibly expressing the Pro291fsinsC-HNF1A mutation, which acts to suppress endogenous HNF1A function, and identified two miRNAs, miR-103 and miR-224, which were significantly elevated upon suppression of endogenous HNF1A function. Furthermore, we found that both miRNAs were potently elevated, in the serum of HNF1A-MODY patients when compared with family controls (mean levels 47- and 293-fold higher, respectively).
Several microRNAs have recently been suggested to play important roles in the pathophysiology of diabetes [13, 14]. miR-103, which we found to be upregulated in INS-1 cells inducibly expressing the Pro291fsinsC-HNF1A mutation, represents a well-characterised miRNA in the context of diabetes and obesity in animal models. miR-103 has been implicated in adipocyte differentiation [28], and was found to be significantly upregulated in the livers of hyperglycaemic rats [29] and mouse models of diabetes [14]. Expression of miR-103 and miR-107 (which differs from miR-103 by a single nucleotide) was also found to be upregulated in obese mice [14], with inhibition of miR-103/-107 leading to improved glucose homeostasis and insulin sensitivity. Caveolin-1, an important regulator of the insulin receptor gene, has been identified as a functional gene target for miR-103 [14]. In the present study, we found elevated miR-103 levels in the serum of HNF1A-MODY carriers. In line with the above-mentioned animal studies, it is possible that hyperglycaemia may be sufficient to contribute to the increase in miR-103 levels in individuals with HNF1A-MODY. However, we did not observe a significant correlation between miR-103 levels and glycaemic control in these individuals. Furthermore, miR-103 was induced in INS-1 cells compared with non-induced control cells when both were cultured at identical glucose levels. These findings therefore suggest that factors other than hyperglycaemia, but triggered by the suppression of endogenous HNF1A function, contributed to miR-103 induction.
miR-224, which we also found to be upregulated in INS-1 cells inducibly expressing the Pro291fsinsC-HNF1A mutation, has not yet been associated with diabetes. It has, however, been previously found to be dysregulated in several types of human cancer, including hepatocellular carcinoma, pancreatic ductal adenocarcinoma and renal cell carcinoma [30–32]. Of note, HNF1A is highly expressed in the organs affected and somatic mutations in HNF1A have been previously associated with these and other cancers [33–35]. Furthermore, the progression of hepatocellular carcinomas in mice has been shown to be associated with a downregulation of HNF1A function [36]. miR-224 levels have been shown to be regulated by the TGF-β/Smad pathway [37], while the TGF-β signalling pathway was predicted to be targeted by miR-224 (ESM Table 1). Additional work is required to identify gene targets regulated by miR-224 in beta cells and to determine their role in HNF1A-MODY and diabetes.
There have been recent studies investigating miRNA expression profiles in individuals with type 1 and type 2 diabetes mellitus. Zampetaki et al identified five particular miRNAs that were found to be elevated 0.3- to 1.0-fold in individuals with type 2 diabetes mellitus [38]. Similarly, a study profiling individuals newly diagnosed with type 1 diabetes mellitus compared with healthy controls revealed 12 human miRNAs that were upregulated approximately twofold when compared with controls. Interestingly, several of these miRNAs were linked to apoptosis regulation (miR-181a, miR-24, miR-25, miR-210 and miR-26a) [39]. Only one miRNA (miR-24) overlapped both the type 1 and type 2 diabetes mellitus groups studied. Our study demonstrated a significant, 47- and 293-fold higher difference in mean miRNA expression for miR-103 and miR-224 when comparing individuals with HNF1A-MODY and their negative controls. We also identified a sixfold difference in mean miRNA expression for miR-103 between HNF1A-MODY carriers and individuals with type 2 diabetes mellitus. Interestingly, the miRNAs previously identified in the above studies did not include miR-103 or miR-224, suggesting that these may be specific to HNF1A-MODY, or may only be found in subpopulations of patients with type 1 or type 2 diabetes mellitus. Further clinical studies are required to test this hypothesis. Over the years a variety of potential biomarkers have been investigated to differentiate HNF1A-MODY from other forms of diabetes, particularly type 1 diabetes and type 2 diabetes. Recently published articles have used findings from genome-wide association studies to develop the potential biomarkers high-sensitivity c-reactive protein (hsCRP) and glycan [40, 41]. However, there are limitations to the clinical use of these biomarkers, including concurrent inflammation in the case of hsCRP and the lack of availability of a high-throughput assay for glycan profiling. Although we provide first clinical proof-of-concept that miR-103 may distinguish HNF1A-MODY carriers from individuals with type 2 diabetes (p = 0.0001), further clinical studies on larger patient cohorts and incorporating other biomarkers, such as hsCRP, are warranted. Nevertheless, our study shows that analysing miRNA levels in serum may be clinically feasible and a promising area for identifying potential biomarkers for HNF1A-MODY. Despite the existence of RNases, miRNAs remain stable in serum and other body fluids, and appear to have the potential to serve as paracrine signalling molecules [42]. The reason for this appears to be controversial, with some arguing that miRNAs are engulfed into lipid or lipoprotein complexes such as exosomes or microvesicles [43, 44]. Others suggest that the export of miRNAs by mammalian cells is independent from exosomes [45] and are in fact derivatives of dead cells that remain bound to Ago2 protein (a part of RNA-induced silencing complex) in the extracellular matrix [46]. Irrespectively, their stability may make miRNAs suitable for biomarker development.
We currently cannot determine whether the miRNAs detected in the serum of HNF1A-MODY carriers originated from beta cells or from other HNF1A-expressing organs such as liver and kidney. Of note, we also found that HNF1A-MODY carriers with high levels of circulating miR-224 frequently showed low levels of miR-103, and vice versa. It is possible that this is a functional interaction, or a consequence of the different types of HNF1A mutations, or of different tissues expressing or secreting different miRNAs in vivo. HNF1A-MODY carriers are relatively insulin sensitive; in this current study, miR-224 and the aggregate of miR-103 and miR-224 correlated positively with OGIS, which is an estimate of glucose clearance in a hyperinsulinaemic–euglycaemic clamp. Further prospective clinical studies on larger HNF1A-MODY patient cohorts are required to determine whether serum levels of miR-224 or miR-103 are associated with a lower or higher risk of develo** complications associated with diabetes or whether they may indicate responses to therapeutic intervention. We did not apply corrections for multiple comparisons to the analyses so as not to miss any potential associations with clinical variables in the current population studied.
The currently available therapeutic options for the management of diabetes and its related complications are not completely effective, suggesting that our knowledge of the disease is incomplete. Because of the roles of microRNAs in the fine tuning of physiological processes and their dysregulation in human diseases such as diabetes, study of microRNAs may provide new insights into disease pathogenesis, and may deliver new targets for therapeutic intervention. Indeed, the first miRNA-targeting agent (an inhibitor of miR-122) has already entered human clinical trials for the treatment of hepatitis C virus infection. Our study highlights the role of miRNAs in the pathogenesis of diabetes and their potential as future therapeutic targets and biomarkers.
Abbreviations
- DBP:
-
Diastolic blood pressure
- HNF1A:
-
Transcription factor 1/hepatocyte nuclear factor 1α
- HNF1A-MODY:
-
MODY due to a mutation in HNF1A
- hsCRP:
-
High-sensitivity c-reactive protein
- INS-1:
-
Insulinoma cell line
- IQR:
-
Inter quartile range
- miR, miRNA:
-
MicroRNA
- OGIS:
-
Oral glucose insulin sensitivity
- RQ:
-
Relative expression
- SBP:
-
Systolic blood pressure
- TGF-β:
-
Transforming growth factor-β
References
Bell GI, Polonsky KS (2001) Diabetes mellitus and genetically programmed defects in beta-cell function. Nature 414:788–791
Kaisaki PJ, Menzel S, Lindner T et al (1997) Mutations in the hepatocyte nuclear factor-1alpha gene in MODY and early-onset NIDDM: evidence for a mutational hotspot in exon 4. Diabetes 46:528–535
Wang H, Antinozzi PA, Hagenfeldt KA, Maechler P, Wollheim CB (2000) Molecular targets of a human HNF1 alpha mutation responsible for pancreatic beta-cell dysfunction. EMBO J 19:4257–4264
Yamagata K, Furuta H, Oda N et al (1996) Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature 384:458–460
Boutin P, Chevre JC, Hani EH et al (1997) An automated fluorescent single-strand conformation polymorphism technique for screening mutations in the hepatocyte nuclear factor-1alpha gene (maturity-onset diabetes of the young). Diabetes 46:2108–2109
Servitja JM, Pignatelli M, Maestro MA et al (2009) Hnf1alpha (MODY3) controls tissue-specific transcriptional programs and exerts opposed effects on cell growth in pancreatic islets and liver. Mol Cell Biol 29:2945–2959
Yamagata K, Nammo T, Moriwaki M et al (2002) Overexpression of dominant-negative mutant hepatocyte nuclear factor-1 alpha in pancreatic beta-cells causes abnormal islet architecture with decreased expression of E-cadherin, reduced beta-cell proliferation, and diabetes. Diabetes 51:114–123
Bonner C, Bacon S, Concannon CG et al (2010) INS-1 cells undergoing caspase-dependent apoptosis enhance the regenerative capacity of neighboring cells. Diabetes 59:2799–2808
Bonner C, Farrelly AM, Concannon CG et al (2011) Bone morphogenetic protein 3 controls insulin gene expression and is down-regulated in INS-1 cells inducibly expressing a hepatocyte nuclear factor 1A-maturity-onset diabetes of the young mutation. J Biol Chem 286:25719–25728
Wang H, Maechler P, Hagenfeldt KA, Wollheim CB (1998) Dominant-negative suppression of HNF-1alpha function results in defective insulin gene transcription and impaired metabolism-secretion coupling in a pancreatic beta-cell line. EMBO J 17:6701–6713
Nammo T, Yamagata K, Hamaoka R et al (2002) Expression profile of MODY3/HNF-1alpha protein in the develo** mouse pancreas. Diabetologia 45:1142–1153
Calin GA, Croce CM (2006) MicroRNAs and chromosomal abnormalities in cancer cells. Oncogene 25:6202–6210
Poy MN, Eliasson L, Krutzfeldt J et al (2004) A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432:226–230
Trajkovski M, Hausser J, Soutschek J et al (2011) MicroRNAs 103 and 107 regulate insulin sensitivity. Nature 474:649–653
Poy MN, Spranger M, Stoffel M (2007) microRNAs and the regulation of glucose and lipid metabolism. Diabetes Obes Metab 9(Suppl 2):67–73
Guo G, Wang W, Bradley A (2004) Mismatch repair genes identified using genetic screens in Blm-deficient embryonic stem cells. Nature 429:891–895
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Kloosterman WP, Lagendijk AK, Ketting RF, Moulton JD, Plasterk RH (2007) Targeted inhibition of miRNA maturation with morpholinos reveals a role for miR-375 in pancreatic islet development. PLoS Biol 5:e203
Baroukh N, Ravier MA, Loder MK et al (2007) MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J Biol Chem 282:19575–19588
Wobser H, Dussmann H, Kogel D et al (2002) Dominant-negative suppression of HNF-1 alpha results in mitochondrial dysfunction, INS-1 cell apoptosis, and increased sensitivity to ceramide-, but not to high glucose-induced cell death. J Biol Chem 277:6413–6421
Asfari M, Janjic D, Meda P, Li G, Halban PA, Wollheim CB (1992) Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130:167–178
da Huang W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Dennis G Jr, Sherman BT, Hosack DA et al (2003) DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4:P3
Kyithar MP, Bacon S, Pannu KK et al (2011) Identification of HNF1A-MODY and HNF4A-MODY in Irish families: phenotypic characteristics and therapeutic implications. Diabetes Metab 37:512–519
Ellard S, Colclough K (2006) Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young. Hum Mutat 27:854–869
Mari A, Pacini G, Murphy E, Ludvik B, Nolan JJ (2001) A model-based method for assessing insulin sensitivity from the oral glucose tolerance test. Diabetes Care 24:539–548
Kozomara A, Griffiths-Jones S (2011) miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39:D152–D157
Kajimoto K, Naraba H, Iwai N (2006) MicroRNA and 3T3-L1 pre-adipocyte differentiation. RNA 12:1626–1632
Herrera BM, Lockstone HE, Taylor JM et al (2010) Global microRNA expression profiles in insulin target tissues in a spontaneous rat model of type 2 diabetes. Diabetologia 53:1099–1109
Mees ST, Mardin WA, Sielker S et al (2009) Involvement of CD40 targeting miR-224 and miR-486 on the progression of pancreatic ductal adenocarcinomas. Ann Surg Oncol 16:2339–2350
Wang Y, Toh HC, Chow P et al (2012) MicroRNA-224 is up-regulated in hepatocellular carcinoma through epigenetic mechanisms. FASEB J 26:3032–3041
Lichner Z, Mejia-Guerrero S, Ignacak M et al (2012) Pleiotropic action of renal cell carcinoma-dysregulated miRNAs on hypoxia-related signaling pathways. Am J Pathol 180:1675–1687
Jeannot E, Mellottee L, Bioulac-Sage P et al (2010) Spectrum of HNF1A somatic mutations in hepatocellular adenoma differs from that in patients with MODY3 and suggests genotoxic damage. Diabetes 59:1836–1844
Rebouissou S, Vasiliu V, Thomas C et al (2005) Germline hepatocyte nuclear factor 1alpha and 1beta mutations in renal cell carcinomas. Hum Mol Genet 14:603–614
Laurent-Puig P, Plomteux O, Bluteau O et al (2003) Frequent mutations of hepatocyte nuclear factor 1 in colorectal cancer with microsatellite instability. Gastroenterology 124:1311–1314
Lazarevich NL, Cheremnova OA, Varga EV et al (2004) Progression of HCC in mice is associated with a downregulation in the expression of hepatocyte nuclear factors. Hepatology 39:1038–1047
Yao G, Yin M, Lian J et al (2010) MicroRNA-224 is involved in transforming growth factor-beta-mediated mouse granulosa cell proliferation and granulosa cell function by targeting Smad4. Mol Endocrinol 24:540–551
Zampetaki A, Kiechl S, Drozdov I et al (2010) Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ Res 107:810–817
Nielsen LB, Wang C, Sorensen K et al (2012) Circulating levels of microRNA from children with newly diagnosed type 1 diabetes and healthy controls: evidence that miR-25 associates to residual beta-cell function and glycaemic control during disease progression. Exp Diabetes Res 2012:896362
McDonald TJ, Shields BM, Lawry J et al (2011) High-sensitivity CRP discriminates HNF1A-MODY from other subtypes of diabetes. Diabetes Care 34:1860–1862
Thanabalasingham G, Huffman JE, Kattla JJ et al (2013) Mutations in HNF1A result in marked alterations of plasma glycan profile. Diabetes 62:1329–1337
Mitchell PS, Parkin RK, Kroh EM et al (2008) Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A 105:10513–10518
Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9:654–659
Hunter MP, Ismail N, Zhang X et al (2008) Detection of microRNA expression in human peripheral blood microvesicles. PLoS One 3:e3694
Wang K, Zhang S, Weber J, Baxter D, Galas DJ (2010) Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res 38:7248–7259
Turchinovich A, Weiz L, Langheinz A, Burwinkel B (2011) Characterization of extracellular circulating microRNA. Nucleic Acids Res 39:7223–7233
Funding
This study was supported by grants from the Health Research Board (RP/2007/316), the Mater Foundation co-funded by the Medical Research Charity Groups/ Health Research Board, and a Mater College Grant to M. M. Byrne, and by grants from Science Foundation Ireland (08/IN1/1949; 12/TIDA/B2387) and the Health Research Board (RP/2008/14) to J. H. M. Prehn.
Duality of interest
The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement
CB and KCN were responsible for acquisition, analysis and interpretation of the data and writing of the article. SB, MPK, CGC and IMB were responsible for acquisition of the data and the writing and revision of the article. JS performed the statistical analysis and assisted with writing and revision of the article. RLS interpreted data and revised article. JHMP and MMB were responsible for conception of the study, analysis and interpretation of the data and the writing and revision of the manuscript. All authors approved the final version of the article.
Author information
Authors and Affiliations
Corresponding author
Additional information
C. Bonner and K. C. Nyhan contributed equally as first authors.
J. H. M. Prehn and M. M. Byrne share senior authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM Table 1
(PDF 39 kb)
Rights and permissions
About this article
Cite this article
Bonner, C., Nyhan, K.C., Bacon, S. et al. Identification of circulating microRNAs in HNF1A-MODY carriers. Diabetologia 56, 1743–1751 (2013). https://doi.org/10.1007/s00125-013-2939-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-013-2939-4