
Abstract
The neurological diseases primarily include acute injuries, chronic neurodegeneration, and others (e.g., infectious diseases of the central nervous system). Autophagy is a housekee** process responsible for the bulk degradation of misfolded protein aggregates and damaged organelles through the lysosomal machinery. Recent studies have suggested that autophagy, particularly selective autophagy, such as mitophagy, pexophagy, ER-phagy, ribophagy, lipophagy, etc., is closely implicated in neurological diseases. These forms of selective autophagy are controlled by a group of important proteins, including PTEN-induced kinase 1 (PINK1), Parkin, p62, optineurin (OPTN), neighbor of BRCA1 gene 1 (NBR1), and nuclear fragile X mental retardation-interacting protein 1 (NUFIP1). This review highlights the characteristics and underlying mechanisms of different types of selective autophagy, and their implications in various forms of neurological diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neurological diseases
The nervous system is regarded as our body’s command center. any impairment or interruption in the nervous system would induce a dysfunctional physiological state, named neurological diseases [1]. Neurological diseases can be categorized as acute injuries (e.g., ischemic or hemorrhagic stroke, spinal cord injury, and traumatic brain injury), chronic neurodegeneration [e.g., Alzheimer’s disease (AD) and Parkinson’s disease (PD)], and others (e.g., brain tumors, center nervous system infectious disease, etc.) [2, 3]. The underlying molecular mechanisms involve neuronal apoptosis, neuroinflammation, oxidative stress, autophagy, etc. [4, 5]. For example, deposits of massive amyloid-β peptide lead to neuroinflammation and oxidative stress in patients with AD, which finally cause neuronal apoptosis [6,7,8]. Furthermore, growing evidence suggests that mitochondrial dysfunction, redox imbalance, massive deposits of aberrant proteins (i.e., α-synuclein), and damage of the ubiquitin–proteasome system contribute to the pathophysiology of PD [9, 10]. Moreover, stroke, defined as a lack of blood supply to the brain or the presence of blood spreading into the brain and subarachnoid space, would cause dysfunction of the mitochondria, endoplasmic reticulum, peroxisome, etc., further introducing oxidative stress and inflammation, and finally causing cell death [11, 12]. Therefore, strategies and programs to treat neurological diseases could significantly reduce the global burden.
Autophagy
Autophagy was first described by Sam L. Clark Jr. as ‘self-eating’ in 1957, which was confirmed by Christian de Duve who found the debris of intracellular organelle structures within lysosomes [13, 14]. Autophagy can be activated by stress (stroke, trauma, etc.) or nutrient deprivation. The main physiological functions of autophagy not only include degradation or recycling of long-lived proteins, but also the elimination of dysfunctional or broken organelles, such as the mitochondria, peroxisomes, or ribosomes [15]. There are three different types of autophagy reported in mammalian cells according to their method of substrate delivery: macroautophagy, microautophagy, and chaperone-mediated autophagy [16]. On the other hand, autophagy could also be regarded as a non-selective pathway for recycling nutrition, but as a selective way to remove dysfunctional and damaged organelles [17].
Macroautophagy
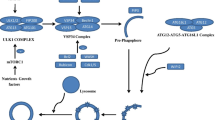
There remains much to be uncovered regarding autophagy-related genetic proteins, and their involvement in different steps of autophagy in yeast, most of which are conserved in mammals (Fig. 1) [18, 19]. The process of autophagy starts with the assembly of phagophore assembly site (PAS). Then, the UNC51-like kinase (ULK) complex assembles to the PAS [20]. After that, the class III phosphatidylinositol 3-kinase (PI3K III) complex helps to form the nucleation of autophagy [21]. Following nucleation, the formation of an Atg5–Atg12–Atg16-like 1 (Atg16L1) complex is required to facilitate cargo recognition and autophagosome membrane elongation [22].

Three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy
Microautophagy
Microautophagy is a catabolic process, in which the dysfunctional or superfluous proteins and organelles are delivered directly to the endosomal/lysosomal lumen (Fig. 1) [23]. However, in mammalian cells, the detailed molecular mechanisms engaged in the process of microautophagy are still not well understood. However, the process of microautophagy largely depends on the endosomal sorting complexes required for transport (ESCRT) I and III systems and the protein chaperone, hsc70 [24].
Chaperone-mediated autophagy (CMA)
The main function of CMA is to degrade the proteins with a KFERQ motif [25]. Nearly 30% of cytosolic proteins contain the KFERQ motif [26]. The lysosome-targeted proteins with a KFERQ motif are first transferred to a lysosome-associated membrane protein 2 (LAMP2A)-containing complex on the lysosomal membrane with the help of chaperones (heat shock cognate 70 (hsc70), heat shock protein 70 (Hsp70)), and co-chaperones, including HSP40, Hsc 70-interacting protein (HIP), Hsp70–Hsp90 Organizing Protein (HOP), Hsp90. Then, the target proteins are unfolded and degraded under the assistance of a complex of proteins in the lysosomal lumen, including Hsc70 [27].
Selective autophagy
Selective autophagy (mitophagy, pexophagy, ER-phagy, ribophagy, and lipophagy) is a process in which a lysosomal-targeted cargo is selectively recognized and degraded, relying on receptor proteins that bind Hsp70–Hsp90 Organizing Protein (HOP) [28] (Fig. 2). Many proteins contribute to the process of selective autophagy. For example, when mitophagy is triggered, Parkin is phosphorylated and activated by PTEN-induced kinase 1 (PINK1), which can further activate and build ubiquitin chains, while autophagy receptors, such as nuclear domain 10 protein 52 (NDP52), SQSTM1 (p62), optineurin (OPTN), and Tax1-Binding Protein 1 (TAX1BP1), target dysfunctional mitochondria to autophagosomal membranes [29, 30]. In pexophagy, peroxin 5 (PEX5) and PEX14 are the peroxisome resident proteins that initiate the pexophagy process [31]. Additionally, selective autophagy is also involved in many pathophysiological processes. For example, the improvement of mitophagy or pexophagy could alleviate the inflammation and oxidative stress after various cellular stresses, and ultimately prevent the cells from dying [32, 33]. The development of neurological diseases leads to the production of many dysfunctional and superfluous organelles (mitochondria, endoplasmic reticulum, peroxisome, etc.), which would introduce severe conditions, such as oxidative stress, neuroinflammation, blood–brain barrier (BBB) disruption, and neuronal apoptosis [34, 35]. Therefore, selective clearance of these organelles is critical to improving neurological functions in patients with neurological diseases.

The autophagy process of five common types of selective autophagy: a the process and regulatory mechanism of mitophagy; b the regulatory mechanism of pexophagy, ER-phagy, ribophagy, and lipophagy
Mitophagy
General introduction of mitochondria and mitophagy
Any disturbance or impairment of the mitochondria would lead to its dysfunction, which would then result in a sharp increase of reactive oxidative species (ROS). Redundant ROS would promote the release of pro-apoptotic factors and finally cause cell death [36, 37]. Mitophagy, the selective degradation of dysfunctional mitochondria defined by Lemasters in 2005, is essential for maintaining cell survival [38]. Furthermore, mitophagy is a process of macroautophagy, and involves three key steps: (1) assembly of phagophore assembly site (PAS); (2) formation of mitophagosome by targeting and engulfment of dysfunctional mitochondria; (3) formation of mitolysosome by fusing with lysosome [39,40,41]. Until now, most of the studies focused on exploring the molecular mechanisms to understand how phagophores are formed, and how dysfunctional mitochondria are recognized.
Many proteins are reportedly involved in the process of mitophagy. One of the most important proteins is PINK1, which is a mitochondrially localized kinase. The main function of PINK1 is to sense the damage of mitochondria, then activate Parkin, and help it translocate from the cytoplasm to the damaged mitochondria [42,43,44,45]. Parkin is an ubiquitin ligase, and normally remains in a “closed” state by hiding the enzyme domain [46]. When mitophagy is triggered, PINK1 will phosphorylate and activate Parkin, which can further activate and build ubiquitin chains. Another group of important proteins is autophagy receptors (Table 1), which assist the autophagy machinery in selectively targeting the mitochondria. These receptors share two important regions to direct mitochondria to autophagy machinery: LC3-interacting regions (LIR) and ubiquitin-binding domains (UBDs). As of now, five autophagy receptors have been reported, including NDP52, OPTN, p62, TAX1BP1, and NBR1 [47, 48]. However, of these five receptors, only NDP52 and OPTN are essential to initiate mitophagy, while others, such as p62, TAX1BP1, and NBR1, have a minor role in mitophagy [49]. NDP52 and OPTN not only target dysfunctional mitochondria to autophagosomal membranes, but also facilitate the formation of autophagosomal membranes by recruiting key factors, including ULK1, double FYVE domain-containing protein (DFCP1), WD Repeat Domain, Phosphoinositide Interacting 1 (WIPI1), etc. [49]. With the exception of the ubiquitination pathway, mitophagy can be initiated by mitophagy receptors, which target damaged mitochondria directly to autophagosomes for further degradation. Mitophagy receptors include NIP3-like protein X (NIX/Bnip3L), BCL2-like 13 (Bcl2L13), BCL2/adenovirus E1B interacting protein 3 (Bnip3), autophagy/Beclin 1 regulator 1 (AMBRA1), FUN14 domain-containing 1 (FUNDC1), and cardiolipid [39, 50, 51].
Mitophagy could be triggered by various stimuli, such as starvation, hypoxia, stroke, or development. Considering the different physiological context of mitophagy, it can be categorized into three different types: basal, programmed, and stress-induced. Basal mitophagy means that the cells would degrade old or abnormal mitochondria under normal physiological conditions [52, 53]. Mitophagy that occurs in different cell types during development is considered ‘Programmed mitophagy’ [54,55,56]. Stress-induced mitophagy refers to the acute degradation of mitochondria as a result of severe extracellular stress [57].
Molecular pathways of mitophagy
Mitophagy pathways are classified as PINK1–Parkin-mediated and Parkin-independent (Fig. 2a).
PINK1–Parkin-mediated mitophagy
PINK1–Parkin-mediated mitophagy depends on the ubiquitination pathway [58], and is initiated with the activation of PINK1. PINK1 functions to sense mitochondrial damage signaling. Normally, PINK1 is delivered into the mitochondria with the help of TOM and TIM complexes, which are the inner and outer membrane translocases [59]. The N-terminal of PINK1, which is located on the inner membrane, would be cleaved by proteases [60,61,62,63], and the C-terminal is released back to the cytosol, and degraded in an N-end manner [64]. Therefore, the successful import of PINK1 maintains PINK1 at reduced activity. However, membrane potential dissipation prevents the importation of PINK1 into the mitochondria, disrupting the stability of PINK1 [57, 65, 66]. Next, PINK1 is activated via autophosphorylation [67,68,69], dimerization [70], and accumulation [59].
In healthy mitochondria, Parkin closes its enzyme domain via intramolecular interaction. To fully activate Parkin, functional PINK1 must complete two important phosphorylation processes, one is S65 in the Ubl domain of Parkin [71, 72] and another is an analogous S65 residue on ubiquitin (referred herein as pUb) [73,74,75]. The phosphorylation by PINK1 changes Parkin’s conformation, facilitates its interaction with mitochondria, and activates its E3 ligase activity [76, 77]. Afterwards, Parkin acts as an ubiquitin enzyme that works on the proteins of the mitochondrial outer membrane. PINK1 phosphorylates Poly-Ub chains, which act as an ‘eat me’ signal for further recognition.
Phosphorylated poly-Ub is recognized by autophagy receptors (p62, OPTN, etc.), which can promote the formation of the autophagosome by binding with LC3. TBK1 reportedly facilitates OPTN binding to Ub chains by phosphorylating OPTN and promotes the efficacy of mitochondrial clearance [78]. Moreover, OPTN and NDP52 can promote the synthesis of autophagosomal membranes through the recruitment of some key components of autophagosome biogenesis (WIPI1, ULK1, and DFCP1) [49]. In a recent study, Abudu et al. showed that NIPSNAP1 (nipsnap homolog 1) and NIPSNAP2, which are considered mitochondrial matrix proteins, act as “eat me” signals for damaged mitochondria to maintain sustained recruitment of SQSTM1-like receptors (SLRs) to ensure efficient mitophagy [79].
Parkin-independent mitophagy
Surmounting evidence showed that other ubiquitin E3 ligases, such as ariadne RBR E3 Ubiquitin Protein Ligase 1 (ARIH1), Gp78, siah E3 Ubiquitin Protein Ligase 1 (SIAH1), and mitochondrial E3 Ubiquitin Protein Ligase 1 (MUL1), participate in promoting mitophagy, with the exception of Parkin [80,81,82,83,84]. These ubiquitin E3 ligases perform their functions by interacting with the mitochondrial membrane, generating ubiquitin chains, and promoting the recruitment of autophagy receptors (OPTN, NDP52, p62, etc.). These receptors then interact directly with LC3 and attach Ub-tagged organelles into autophagosomes [49].
Role of autophagy receptors in mitophagy
Moreover, some ubiquitin-independent mitochondrial proteins, such as BCL2L13, NIX, BNIP3, and FUNDC1, interact directly with LC3 and GABARAP on autophagosomal membrane without ubiquitination, and mediate mitophagy [85]. BCL2L13 is a functional homologue of Atg32 in mammals with an LIR motif, which interacts directly with LC3 to promote mitophagy in a Parkin-independent manner [86]. Besides, other proteins, such as NIX, BNIP3, and FUNDC1, are outer mitochondrial membrane proteins, and act as mitophagy receptors, which mediate mitochondrial clearance in response to different mitochondrial stresses. The NIX plays an especially important role in programmed mitophagy during differentiation [54,55,56, 87]. NIX-deficient cells accumulate mitochondria, leading to increased apoptosis and developmental defects [88]. LIR motif phosphorylation enhances NIX association with LC3 under stress conditions [89]. Although the signaling cascade of NIX-mediated mitophagy is not yet determined, Rheb, a small GTPase, may be involved, as its mitochondrial localization and physical interaction with NIX regulate mitochondrial removal and maintenance of energy metabolism [90].
Different from other receptors, BNIP3 participates in the regulation of mitochondrial dynamics by inducing mitochondrial fission through optic atrophy 1 (OPA1) disassembly and release, and by recruiting dynamin-related protein (DRP1) to mitochondrial outer membrane [91, 92]. BNIP3 mediates PINK1 stabilization by inhibiting its proteolytic cleavage [93]. Both NIX and BNIP3 sustain mitochondrial homeostasis through regulation of Parkin recruitment, suggesting crosstalk between mitophagy receptors and the PINK1–Parkin pathway [94].
FUNDC1 acts as a conserved mitophagy receptor and mediates mitophagy when there is a deficiency in oxygen and blood [95]. FUNDC1 interacts with both fission and fusion machinery components, regulating mitochondrial dynamics. Mitochondrial phosphatase phosphoglycerate mutase 5 (PGAM5) dephosphorylates FUNDC1, thereby disrupting its physical association with OPA1, and inhibiting mitochondrial fusion under hypoxic conditions. In turn, FUNDC1 translocates to ER–mitochondrial contact sites, mediating DRP1 recruitment and mitochondrial fragmentation. Thus, FUNDC1 coordinates mitochondrial morphology and mitophagy under stress. FUNDC1 may also serve as an ULK1 adaptor; their interaction promotes ULK1 relocation on mitochondria, allowing de novo phagophore biogenesis [96].
Taken together, the diverse repertoire of receptor and adaptor molecules highlights the existence of compensatory mechanisms that regulate mitochondrial numbers in response to environmental and/or intracellular signals. The complex interplay between mitophagy pathways ensures energy metabolism and tissue homeostasis. Thus, maintenance of mitochondrial function, through a fine-tuned mitochondrial quality control system, is critical for cellular and organismal survival [97].
Regulation of mitophagy: activation and inhibition
Impaired mitophagy is believed to be a key factor resulting in many pathological conditions. However, overactivation of mitophagy is also harmful for the cell hemostasis [98, 99]. Therefore, maintaining the balance of promotors and inhibitors is quite important for mitochondrial quality and cell hemostasis.
Studies aiming to discover pharmacological reagents capable of promoting the clearance of dysfunctional or damaged organelles are becoming more prevalent [100]. Positive activators of autophagy, such as rapamycin and metformin, regulate the activity of 5′ AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR), and assist in preserving energy metabolism, possibly by balancing mitochondrial clearance and biogenesis [101, 102]. Rapamycin administration reportedly exerted positive effects on regulating mitochondrial quality by maintaining energy homeostasis and stress resistance in mammalian cells [103, 104]. Metformin supplementation triggers mitophagy by increasing the activity of Parkin, and by downregulating P53 levels [105]. In addition, some other natural compounds, such as urolithin A, resveratrol, and antibiotics, also maintain mitochondrial integrity by inducing mitophagy. Moreover, the mitophagy triggered by these compounds exerts protective and anti-aging effects by restoring energy hemostasis in both mammals and yeast [106,107,108,109,110]. PMI (p62–SQSTM1-mediated mitophagy inducer), one type of artificial chemical, can stabilize nuclear factor erythroid 2–related factor 2 (Nrf2) and induce p62-mediated mitophagy [111]. Ubiquitin Specific Peptidase 8 (USP8) is a cytoplasmic deubiquitinating enzyme (DUB), and functions as a promotor of mitophagy [112, 113]. USP8 has no effect on Parkin’s substrates. On the contrary, it deubiquitinates and stabilizes Parkin directly by removing K6-chains from Parkin [113].
In addition to promoting mitophagy, some negative regulators of mitophagy have been determined. As the induction of mitophagy largely depends on ubiquitination (such as Parkin-dependent pathway), a growing number of researchers have placed their emphasis on DUBs to downregulate mitophagy. From the ~ 80 active DUBs discovered in mammal cells [114], USP35, USP30, and USP15 exert direct deubiquitination effects on Parkin substrates, thus negatively regulating mitophagy [115]. Recently, Wang et al. [116] found that PTEN-L could act as an inhibitor of mitophagy by directly dephosphorylating Ub and Parkin.
Taken together, maintaining the balance of mitophagic promotion and inhibition is important for normal mitochondrial functions and cellular homeostasis. Studies attempting to discover new compounds with both biogenic and mitophagic abilities provide promise for develo** novel therapeutic strategies on mitochondrial diseases [117]. Besides, the post-translational modifications for mitophagy can be concluded as ubiquitination/deubiquitination, acetylation/deacetylation, and phosphorylation/dephosphorylation [118].
Pexophagy
General introduction of peroxisome and pexophagy
Peroxisomes are heterogeneous and dynamic organelles, and vary in number, size, and function among different cell types and metabolic status. This versatile organelle primarily functions to degrade fatty acids, such as very-long-chain and polyunsaturated fatty acids, and metabolize reactive oxygen species (ROS) [119,120,121,122]. Peroxisome homeostasis depends on the balance between the degradation and biogenesis of peroxisomes in different physiological contexts. Any disturbance of the integrity and number of peroxisomes would disrupt the homeostasis of cells, leading to cell death. Therefore, the selective removal of superfluous and damaged peroxisomes, known as pexophagy, is critical to maintain redox homeostasis [123].
The term pexophagy was first described by Klionsky in 1997 [124]. Later, researchers found that pexophagy could be divided into two modes, macropexophagy and micropexophagy [125,126,127]. In mammals, macropexophagy means that a single peroxisome is engulfed by autophagosomes to form a pexophagosome, which is fused with lysosomes and then degraded for recycling. In micropexophagy, the peroxisome is engulfed by vacuolar sequestering membranes (VSMs) and micropexophagy-specific apparatus (MIPA) [128], which forms a lid over the cup-shaped VSMs cradling the peroxisomes [129].
Except for the proteins of core autophagy machinery, many specific proteins are reportedly involved in the process of pexophagy, such as autophagy receptors (Fig. 2b). In mammalian cells, the NBR1 and SQSTM1/p62 are reportedly the autophagy receptors for pexophagy [130]. These receptors share two similar functional domains. For example, LIR binds to LC3, and thus delivers peroxisome to the autophagosome. The other is a ubiquitin-associated domain that allows itself to interact with ubiquitinated residues on the peroxisome [131]. Although SQSTM1 plays an important role in pexophagy, it is not required for pexophagy when NBR1 is sufficient. However, SQSTM1 can increase the efficiency of NBR1-mediated pexophagy by binding with NBR1 [132]. Additionally, these two receptors are not only specific for pexophagy, they are also reported to participate in mitophagy, lysophagy, and ER-phagy [133,134,135]. Besides, PEX14 is reportedly involved in the pexophagic process by directly interacting with LC3-II under nutrient starvation [136]. NBR1 and/or SQSTM1/p62 reportedly facilitate the interaction between PEX14 and LC3-II by inducing a conformational alteration of PEX14, which allows LC3-II to interact with the transmembrane domain of PEX14 [137]. Recent studies show that PEX5 ubiquitination is an important mechanism in initiating pexophagy in response to some stresses, such as peroxisomal dysfunction or oxidative stress. Furthermore, another important factor is ataxia-telangiectasia mutated (ATM) kinase. Activation of ATM could phosphorylate and activate PEX5, which leads to PEX5 self-ubiquitination and pexophagic promotion [138, 139].
Molecular mechanisms of pexophagy
Ubiquitination-mediated pexophagy
Growing evidence has shown that ubiquitination of some specific proteins is the requisite for selective autophagy [45, 140, 141]. Recently, PEX5 ubiquitination is found to be a key role in the pexophagy. Oxidative stress signaling phosphorylates and activates peroxisome-localized ATM, which activates PEX5 via phosphorylation at S141. Then, phosphorylated PEX5 can be ubiquitinated at K209 by the peroxisomal E3-ligases PEX2/10/12 and recognized by SQSTM1/p62, which targets peroxisomes for pexophagy [138].
Adaptor-mediated pexophagy
SQSTM1/p62 acts as an autophagy adaptor and has two important functional domains: LIR of the motif and a UBA domain at the C-terminus [134, 142]. As an autophagy adaptor, SQSTM1/p62 is the regulatory center for autophagic signaling pathways, and is always adapted as a biomarker for evaluating the level of autophagy [134, 143, 144]. In the process of pexophagy, the LC3-interacting region (LIR) of SQSTM1/p62 binds with LC3-II, and the Ubiquitin-Associated (UBA) domain connects with ubiquitinated regions of peroxisomes, resulting in pexophagy and engulfment of the peroxisome [145, 146].
NBR1 is another mediator for pexophagy. NBR1 also contains LIR at the center of the protein and a UBA domain at the C-terminus [147, 148]. NBR1 upregulation reportedly promotes pexophagy by recruiting peroxisomes and acting as a “see me” signal to be recognized by lysosomes [132]. Besides, p62 lacks a juxta-UBA (JUBA) domain that is required for subcellular localization, but it can promote the efficacy of NBR1-induced pexophagy by interacting with NBR1 [132].
ER-phagy (reticulophagy)
The homeostasis maintained by ER is vital for both cellular activity and cell survival. Various exogenous or intracellular stresses, such as imbalance of calcium flux, oxidative stress, protein-folding dysfunction, and disruption in ER functions, which leads to the accumulation of unfolded or misfolded proteins, causing ‘ER stress’ [149,150,151]. One salvage measure that responds to the ER stress is the selective degradation of misfolding proteins or the ER membrane itself. The term “ER-phagy”, also known as “reticulophagy”, was first described by Bernales et al. in 2007, who also found that ER-phagy was induced by ER stress [152].
Many researchers, inspired by the study of mitophagy and pexophagy, have sought to discover the autophagy adaptors or ER-phagy receptors (Fig. 2b). Recently, family with sequence similarity 134, member B (FAM134B) was reported to show advantages in facilitating the degradation of ER membranes [153]. FAM134B, an intramembranal ER-resident protein, is characterized by the presence of a reticulon homology domain (RHD). Khaminets et al. found that the FAM134 reticulon protein family could act as receptors to interact with LC3 or gamma-aminobutyric acid receptor-associated protein (GABARAP), and promote the turnover of ER membrane (‘ER-phagy’). Reticulon 3 (RTN3), an RHD-containing protein, is located at ER tubules, and its major function is to facilitate the formation of ER tubules [154]. Among several splicing isoforms of RTN3, only the longest one is equipped with six LIR domains, which could bind LC3/GABARAP, promote the segmentation of ER tubules, and finally lead to ER-phagy. In fact, RTN3 initiates ER-phagy mainly under conditions of energy or oxygen deprivation. However, RTN3 and FAM134B only exert their function as an ER-phagy receptor in the region they are located [155]. Additionally, a specialized ER-phagy receptor, cell-cycle progression 1 (CCPG1), was discovered, and acts in response to the massive accumulation of misfolded or aggregated proteins in the ER. CCPG1 is a transmembrane protein, and resides in the ER. In yeast, it can prevent cells from cell-cycle arrest, hence the name [156]. As an autophagy receptor, CCPG1 has an LIR motif, which binds with LC3. Moreover, CCPG1 also has an FIR motif that interacts with autophagic proteins, RB1CC1/FIP200. Binding to RB1CC1/FIP200 increases the efficiency of ER-phagy [157]. Recently, Chen et al. have identified a new ER-phagy receptor, Atlastin GTPase 3 (ATL3), which belongs to a family of dynamin-like GTPase. ATL3 has been shown to facilitate ER fusion. Besides, as an ER-phagy receptor, especially for tubular ER, ATL3 binds with GABARAP subfamily proteins through 2 GABARAP-interacting motifs (GIMs) [158].
Unlike other forms of selective autophagy, ER-phagy has its own characteristics. First, most of the ER-phagy receptors are ER-resident proteins. For example, FAM134B [153], the first ER-receptor identified in mammalian cells, is an intramembrane ER-resident protein which has an RHD at the N-terminal. Moreover, FAM134B possesses an LIR domain at C-terminal, which can bind to GABARAP/LC3. ATL3, a recently identified example of an ER-phagy receptor, has two transmembrane regions. This differs from other autophagy receptors, because the LIRs of ATL3 are specific GIMs, which can specifically bind to the GABARAP subfamily. Second, only some portions or fragments of the ER are involved in the selective degradation process, but not the entire organelle. Additionally, in other types of selective autophagy, such as mitophagy or pexophagy, the cargoes are wholly encapsulated by the autophagosome [159]. Thirdly, receptors mediate the selective autophagy of different sub-regions of the ER. For example, FAM134B primarily targets sheet-like ER for degradation, while the long isoform of RTN3 (RTN3L) and ATL3 has effects on tubular ER [153, 155, 160]. Fourth, different ER-phagy receptors exert their functions in different pathophysiological contexts. For example, RTN3, FAM134B, and ATL3 are activated by nutritional deficiencies, while CCPG1 is activated by ER stress. Finally, different ER-phagy receptors mediate ER-phagy in different cell types or tissues. For instance, FAM134B is located in embryonic fibroblasts and U2OS cells, and RTN3L is expressed in kidney and heart, while ATL3 mainly exert its functions in the absence of RTN3L [161, 162].
The ER-phagy shows its physiological value in two ways: (1) some parts of the ER, which are dysfunctional or have accumulated unfolded or misfolded proteins, could be engulfed and degraded via ER-phagy; (2) ER-phagy may represent an important response to ER stress [163].
Aggrephagy
Protein aggregation means the accumulation of unfolded or misfolded proteins, which twine together to form insoluble clumps. The aggregates are always detrimental to the cells, and cause a series of pathological problems, including AD, PD, etc. [164,165,166]. To be noted, aggregation reportedly acts in a protective role for the cell by isolating damaged or dysfunctional proteins in an insoluble form [167]. In cells, three systems are responsible for the quality of proteins: chaperone-assisted folding, proteasomal-dependent degradation, and aggrephagy, a form of selective autophagy that participates in the degradation of ubiquitinated aggregates [168]. These proteins are labeled with ubiquitin (Ub), which binds to their adaptors. The subsequent process of aggrephagy can be briefly explained by the damaged or unfolded proteins that form aggregates, which is labeled by ubiquitination. Then, the aggregates are recognized and engulfed by a double-membrane to form autophagosomes. Finally, the autophagosomes fuse with lysosomes for further degradation and recycling [169].
Like other forms of selective autophagy, aggrephagy depends on the functions of adaptors, such as SQSTM/p62, NBR1, histone deacetylase 6 (HDAC6), and ALFY (autophagy-linked FYVE domain-containing protein) [170,171,172,173]. Ubiquitination of misfolded proteins is a key mediator in the recognition and degradation of protein aggregates by aggrephagy. Since all of these receptors possess one or more LC3 interaction regions (LIRs) and one ubiquitin-binding domain (UBA), the proposed role of these receptors in aggrephagy is to bridge LC3/GABARAP family members with ubiquitinated substrates [174]. In both p62 and NBR1, the UBD that is located in the C-terminal region specifically recognizes Lys63-linked polyubiquitin substrates and forms a complex [175]. Simultaneously, the LC3-interacting motif in p62 and NBR1 promotes the delivery of complexes to form autophagosomes. Among these receptors, p62 is the only essential one for the regulation of substrate ubiquitination [176]. Additionally, p62 recruits a 400 KD autophagy-linked FYVE (ALFY) nuclear protein into the cytoplasm for autophagic degradation of aggregates. ALFY is crucial in facilitating interaction between p62-linked aggregates and the membrane-bound autophagosome, LC3 [177]. The ALFY C-terminal region has BEACH, FYVE, and WD40 domains, which are crucial to this peptide’s functional role in aggrephagy [178]. In particular, the binding of its WD40 domain to Atg5 is essential for ATG5–ATG12–ATG16L1 E3 ligase complex formation. Binding of its FYVE domain to PtdIns3P enhances phagophore formation, while its BEACH domain binds to the p62-aggregate complex and acts as a scaffold between LC3 in phagophores [173]. Under normal autophagy conditions, p62 and NBR1 aggrephagy receptors facilitate aggregate degradation. Tripartite Motif Containing 50 (TRIM50) is an E3 ubiquitin ligase, and it reportedly increases the aggregation of polyubiquitinated substrates in aggresomes. It enhances aggrephagy by increasing p62 expression and by influencing HDAC6-mediated misfolded protein retrograde axonal transport when proteasomal aggregate degradation is impaired [179]. Misfolded proteins generated in axons and dendrites are retrogradely transported to the lysosome-rich microtubule-organizing center (MTOC). In MTOC, they are packed into aggresomes, and are subsequently degraded in the lysosome [180]. These functions are regulated by histone deacetylase-6 (HDAC6). HDAC6 is a deacetylating enzyme that is crucial in microtubule transport machinery [181]. In aggrephagy, HDAC6 deacetylates α-tubulin, cortactin, and HSP-90 [182]. Furthermore, HDAC6 is actively involved in the sorting of polyubiquitinated misfolded proteins for the axonal retrograde transportation that uses Dynein-snapin, a motor–adaptor complex [183].
Others
Selective clearance of ribosome is known as ribophagy, which was first noted by Kraft et al. in 2008. They found that in the setting of nitrogen starvation, the components that formed the 60S subunit of ribosome are more likely to undergo degradation in a lysosomal-dependent manner than the control cytoplasmic proteins [184, 185]. Ubp3- and Bre5-dependent degradation of ribosomes has been observed in yeast upon starvation. In addition, Kraft et al. have reported that Rsp5 was also involved in the regulation of ribophagy; however, it was not essential. Ribophagy has recently been identified in mammalian cells by Wyant et al. [186]. Indeed, their report has revealed the presence of a putative ribosome receptor-NUFIP1, which is required for ribophagy. NUFIP1 contains an LIR motif, and it can directly interact with LC3, thereby delivering ribosome to the lysosome for degradation. However, the ribosomal factor, which is recognized by NUFIP1, has not yet been identified. Therefore, more studies are necessary to focus on exploring the underlying mechanisms of ribophagy induction and its regulatory pathway.
Recently, it was reported that the LD can also be degraded in a lysosome-dependent pathway, known as lipophagy [187]. Lipophagy refers to a process in which LDs are isolated and engulfed by an autophagosome, which then fuses with lysosomes to be degraded [188,189,190]. Interestingly, given that the volume of LDs is much larger (almost 200 μm) than that of lysosomes (0.1–1 μm), the autophagosome membrane always forms on the LDs surface and pinches off parts of the LD membrane to form autolysosomes [191]. Although it is regulated by hypothalamic metabolic neurons [192], as well as many other proteins, the detailed mechanisms of lipophagy regulation remain unclear. Besides, what is currently known, is that lipophagy is quite important for cellular energy metabolism.
Moreover, the lysosomal system is the major organelle to receive cargo from the phagocytic, autophagic, and endocytic pathways, and plays an important role in maintaining nutrient and energy homeostasis. Therefore, the normal function of the lysosomal system is quite important for cellular homeostasis [193]. Several studies have demonstrated the detrimental damage incurred by a dysfunctional lysosomal system, rupture of lysosome membranes in neurodegenerative disorders, infectious diseases, and tumors. Limited damages can be repaired by the endosomal sorting complex required for transport (ESCRT) machinery. Otherwise, the lysosomes will be ubiquitinated and degraded by selective macroautophagy, known as lysophagy [194, 195]. Currently, growing studies showed that the stress-induced exposure of luminal glycans to the cytosol is the critical factor to induce ubiquitination [196]. The recognition and encapsulation of ubiquitinated lysosomes by the double-membrane depends on the functions of adaptors, such as SQSTM1/p62, TAX1BP1, and NDP52 [197, 198].
Selective autophagy in neurological diseases: friends or foes?
There is growing evidence to suggest that there is a close relation between selective autophagy and neurological diseases (Table 2). Until now, aggrephagy and mitophagy have been the most intensively investigated types of selective autophagy in neurological diseases, whereas other types are reported less. The protective or injurious effects of selective autophagy in the occurrence and development of CNS diseases differ from each other. Similarly, the role of selective autophagy in different stages during the development of CNS diseases is different, as well. Consequently, overactive or insufficient selective autophagy may damage cells. Therefore, determining how to show the protective effect of selective autophagy on tissues (cells), and avoiding or reducing its damage to tissues (cells) to the greatest extent will likely be the focus of future research by scholars.
Parkinson’s disease
Parkinson’s disease is a neurodegenerative disease characterized by the degenerative loss of dopaminergic neurons in the pars compacta of the substantia nigra (SNpc). Its main pathological feature is the formation of eosinophilic inclusion bodies (Lewy bodies) in neurons, which are mainly composed of α-nuclear synaptic proteins [199]. The Lewy bodies are more likely to form in aggregates due to genetic variation in both types of PD, which finally disrupts the cellular homeostasis, leading to pathology [200, 201]. Moreover, cell-to-cell propagation of malformed α-synuclein causes the contamination of healthy cells [202]. Finally, patients manifest with typical motor disturbance once the pathology causes the loss of more than 50% of dopaminergic neurons in the SNpc [203]. Once chaperone-mediated or proteasomal-dependent mechanisms fail to clear the aggregates, selective autophagy can be an alternative way to clear them [204]. Pharmacological promotion of autophagy reportedly shows neuroprotective effects for PD via selective clearance of α-synuclein aggregates [205,206,207]. For the regulation of aggrephagy in PD, mutations in DJ-1 or alpha-synuclein (SNCA) have been indicated to suppress aggrephagy, whereas the upregulation of NBR1 and p62 promotes aggrephagy [208]. Another recent study has shown that estrogen-related receptor α (ERRα) also participates in aggrephagy by restraining autophagy flux [209]. Besides, several studies show that mitochondrial dysfunction plays a key role in the pathogenesis of sporadic PD. In the sample of PDs, researchers found that dysfunction of electron transport complexes exists in almost 25% of patients with sporadic PD [210]. As a result, some agents, such as opiate analogue 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and pesticides, can disrupt the function of the electron transport chain (ETC) and trigger a Parkinsonian phenotype in the models [211, 212]. Previous reports indicated that mitochondria within the SNpc neurons are more likely to be influenced by various stresses and damages. Therefore, any mitochondrial damage and accumulation of the damaged mitochondria cause progressive damage to the mitochondria along the whole life, unless the damaged mitochondria are labeled and selectively degraded via mitophagy. The genes encoding for α-synuclein, PINK1, DJ-1, and Parkin are all involved in mitophagy, which selectively degrade the heavily damaged mitochondria, thereby avoiding their toxic accumulation [213, 214]. Consequently, any disturbance in the genetic expressions leads to mitophagic dysfunction, further causing neurodegeneration [215]. Three mechanisms are reportedly responsible for initiating mitophagy: ubiquitin-mediated, cardiolipin-mediated, and transmembrane receptor-mediated [47]. Oh et al. have reported that S-nitrosylated PINK1 (SNO-PINK1) can impair mitophagy, whereas mitochondrial insults stimulated by age- or environmental-related stresses lead to the increase of SNO-PINK1, which inhibits the activity of SNO-PINK1. Hence, the formation of SNO-PINK1 and functional disturbance of mitophagy greatly lead to the pathogenesis of PD [215]. Besides, DNA damage is very common in the progression of PD. Accumulated DNA damage leads to a series of biochemical cascades, ultimately resulting in cellular outcomes, such as mitophagy or cell death [216]. One possible way is through the ATM–AMPK axis. ATM, a master regulator of the DNA damage response, can either directly phosphorylate or activate AMPK, which can further promote mitophagy through phosphorylation [217]. Similarly, Chen et al. [158] revealed that ATL3 is the receptor and promotor of ER-phagy, as well as the mediator of ER fusion through its specific binding to GABARAP subfamily proteins, which suggests the potential role of ER-phagy in neurodegenerative diseases.
Alzheimer’s disease
Alzheimer’s disease (AD), a type of neurodegenerative diseases, is the major reason of dementia worldwide. The main clinical manifestations of AD typically begin with memory loss, but later presents with defects in cognitive and adaptive functioning [218,219,220]. AD is pathologically characterized by neuronal loss, intracellular deposits of hyper-phosphorylated tau protein, and the accumulation of amyloid-β (Aβ) in cerebral vasculature and brain parenchyma [221]. In addition, emerging evidence indicates that dysfunctional ER and mitochondria play a key role in the pathogenesis of AD [222,223,224].
The underlying molecular mechanisms of AD are still far from understood, but aggrephagy disturbance was reported to be a critical mechanism of AD. For example, the autophagy–lysosome pathway has been underlined as an important point for Aβ clearance [225]. In normal human CNS, there is no Aβ protein accumulation due to the higher rate of clearance compared to production, which reveals the critical role of autophagy for the degradation and production of Aβ protein [226,227,228]. Moreover, growing evidence suggests that there is dysfunction of the autophagy–lysosomal system in early stages of AD (without accumulation of neurofibrillary tangles or Aβ proteins), and thus, normal maintenance of autophagy has been considered a promising regimen for treating AD [228].
It is well established that mitophagy is critical for degrading dysfunctional mitochondria and maintaining mitochondrial homeostasis, a critical process for normal neuronal function. Conversely, defects in mitophagy lead to AD [229,230,231]. In line with these, several studies have reported that dysfunctional and damaged mitochondria were found in the brain tissues from both AD patients and animal models [229, 232]. A deficiency in mitophagic activity has been shown in a Presenilin1 mutant AD model, which directly shows the potential role of mitophagy in AD [233]. Experimental induced mitochondria dysfunction using treatment with agents or genetic alteration aggravates the manifestation of AD by increasing the deposit of Aβ and pTau aggregation [234,235,236,237]. The inhibitory role of mitophagy in the accumulation of Aβ and tau proteins has been reported by Fang et al., resulting in reversed cognitive deficits in models of AD. Resultantly, impaired removal of defective mitochondria has been suggested to be a critical factor in the pathogenesis of AD, indicating that mitophagy may be a promising therapeutic strategy [230]. Besides, it was reported that mitophagy [238] and DNA damage [239] have been closely associated with the development and progression of AD [240]. AD also shows a reduction in base excision repair (BER) [241] and double-strand breaks (DSB) [242] repair, which can be sensed by DNA-damaged sensors, such as ATM or cGAS-STING pathway, which further activates mitophagy by modulating its downstream targets [243].
Similar to PD, aggrephagy has an important role in clearing the abnormal proteins found in AD. It was reported that decreased Beclin1, defects in the lysosomal system, massive deposits of Aβ or tau proteins, and phosphorylation of P62 all decrease the effects of aggrephagy and contribute to AD [244,245,246,247,248,249].
As mentioned above, any stress causing the massive deposit of abnormal proteins in the ER lumen causes ER stress. The cells increase its protein-folding ability to deal with mild ER stress, but once this fails, the cells turn to autophagy for help [250, 251]. The earliest evidence that ER participates in autophagy was from a report, suggesting that ER acts as the source of autophagosome membrane [152]. However, ER can also be degraded by autophagy. The induction of severe ER-stress activates selective autophagy of ER, known as ER-phagy [152, 252]. The main function of ER-phagy is to isolate and degrade some parts of the ER with abnormal aggregates, which cannot be handled by other methods. What is the relationship between ER-phagy and AD? Accumulating evidence has shown that UPR activation markers are extensively increased in the brain tissues from AD patients and animal models [253, 254]. Lai et al. [255] also indicated that deposition of Aβ proteins inhibits the interaction between ER and microtubules in the hippocampal neurons, which causes the dysfunction of ER and activation of the lysosomal–autophagy system. Besides, chemicals that interfere with cholesterol metabolism within the ER reportedly increase the efficiency of Aβ42 clearance by autophagy, further indicating the close relationship between ER dysfunction and autophagy in AD [256]. Moreover, initiation of ER stress by with agents triggers autophagy, and greatly decreases the amount of mature APP and amyloid beta precursor-like protein 1 (APLP1), while autophagic inhibition contributes to the deposition of amyloid precursor protein (APP) protein and APLP1 [257]. In addition, tau protein also assists in understanding the relationship of ER and autophagy in AD. Loewen and Feany [258] have found that induction of ER stress can sometimes reduce the harmful effects of tau via introduction of autophagy.
LDs are abundant in the neurons. They mainly include glycerophospholipids, sphingolipids, and cholesterol. The normal metabolism of these lipids is quite important for the maintenance of neuronal functions. For example, cholesterol forms the main components of cell membranes and myelin, which is critical for synapse, dendrite formation [259, 260], and axonal guidance [261]. Several studies show that decreased cholesterol in neurons greatly affects neuronal activity and neurotransmission, causing the progressive degeneration of dendritic spine and synapse, which contributes greatly to the pathogenesis of AD [262,263,264]. Besides, sphingomyelinases have been shown to increase neuronal apoptosis by generating the pro-apoptotic molecule, ceramide [265, 266]. Besides, the level of arachidonic acid increases in the brain tissue from AD model [266, 267]. Therefore, selective autophagy of dysfunctional lipids is another key therapeutic target in treating AD. Importantly, although enhancing lipophagy has previously been indicated to reduce the accumulation of lipid droplets, and thus decrease neuronal neurodegeneration caused by the accumulation of dihydroceramide desaturases [268], there is a lack of literary evidence supporting the direct relation between lipophagy and AD. Therefore, future studies are warranted to understand the relationship between lipophagy and AD.
Stroke
Stroke, one of the most common types of neurological diseases, is an acute cerebrovascular incident caused by either a sudden rupture of blood vessels feeding the brain (hemorrhagic stroke) or a failure of blood flow into the brain due to an abrupt blockage of blood vessels (ischemic stroke) [269]. Mitochondrial dysfunction contributes greatly to brain injury after stroke, as the mitochondria are the energy suppliers and important organelles in the regulation of oxidative metabolism and cellular apoptosis. As mentioned above, mitophagy is responsible for controlling the number and quality of mitochondria by degrading damaged and accumulated mitochondria. Besides, mitophagy is also important for maintaining the physiological functions of mitochondria, such as mitochondrial fusion and fission, or oxidative metabolism. However, the exact molecular mechanisms regarding the involvement of mitophagy in stroke remain unclarified [270]. During ischemic stroke, mitophagy is critical for maintaining normal function of the mitochondria, while aggressive mitophagy leads to cell death. Shi et al. [98] have reported that NIX mainly controls the basal level of mitophagy in physiological conditions, while BNIP3 can induce excessive mitophagy. The contribution of 12/15-lipoxygenase (LOX) to the disease pathogenesis through the increase of oxidative stress-related injury has been implicated, particularly in stroke. Besides, 12/15-LOX knockout has been shown to lead to increased macroautophagy, mitophagy, and pexophagy. It is widely accepted that inhibition of LOX provides protective effects in many diseases caused by ischemia or oxidative stress, and has also been proposed to be the culprit behind enhanced macroautophagy in the absence of LOX [271]. Next, γ-aminobutyric acid (GABA), the primary inhibitory neurotransmitter, has been shown to inhibit selective autophagy pathways, as well as mitophagy and pexophagy in yeast through Sch9, which is the homolog of S6K1, a mammalian kinase associated with oxidative stress [272]. Pexophagy, the selective degradation of dysfunctional or superfluous peroxisomes, is another selective type of autophagy that is essential for the maintenance of a balanced cellular redox state. After the stroke attack, the peroxisome population greatly increased, including the dysfunctional ones, and both lead to serious oxidative stress, neuroinflammation, and, ultimately, neuronal death. Therefore, selective autophagy of dysfunctional or superfluous peroxisomes is a key target in alleviating brain injury after stroke. Although Zhu et al. have suggested that pexophagy can exhibit neuroprotective effects by reducing the infarct size after ischemic stroke [273], the particular role of peroxisomes and pexophagy in stroke has been grossly underestimated thus far. Therefore, more studies should be carried out to explore the important role of peroxisomes in stroke.
The association between stroke and lipids, especially triglyceride and low-density lipids (LDLs), has been previously confirmed by prospective studies [274, 275], revealing that high triglyceride and ox-LDL levels substantially increase the risk of death and poor functional outcomes, both before and after stroke. Stroke is associated with a series of pathophysiological consequences, including apoptosis, inflammation, oxidative stress, and disruption in lipid metabolism. The alterations in lipid metabolism influence the amount of fatty acids (FA) and neutral lipid storage [276]. Massive accumulation of LDs is detrimental to homeostasis, as accumulation of neutral triglycerides (TGs) always promotes the metabolism of long-chain FA, which contributes to lipoapoptosis [277, 278]. Besides, oxidative stress increases the metabolism of polyunsaturated FAs (PUFAs), whose products, such as malondialdehyde (MDA), can aggravate oxidative stress [279, 280]. Kirac et al. [281] showed that ischemia and reperfusion in the liver increase lipid-mediated inflammation. Therefore, selective degradation of these types of superfluous and harmful lipids, namely lipophagy, can substantially reduce the brain injuries after stroke. Indeed, ischemic stroke reportedly induced the activation of lipophagy that occurred to effectively remove the lipid excess, modulate the lipid homeostasis, and counteract the intracellular TG overload [282]. However, induced excessive lipophagy can be a doubled-edged sword, because some products of LDs, such as ceramides and PUFA-derived lipid mediators, are detrimental to the adjacent cells [283]. Nevertheless, more research is necessary to determine the exact functions of lipophagy in stroke.
In recent years, ER-stress has been shown to play an important role in the pathogenesis of stroke. Insults that disturb ER function result in ER-stress or ER dysfunction. Hence, selective autophagy of dysfunctional ER can be a promising target to reduce injury caused by stroke. Moreover, selective isolation and engulfment of the ER assist cells in dealing with severe ER stress, even without degradation by vacuolar proteases, suggesting that selective sequestration of the ER is a critical mechanism for the cells to go through ER stress [284]. Interestingly, the study of Carloni et al. [285] found that upregulation of ribophagy exerts neuroprotective effects for animals with neonatal hypoxic-ischemia. This can be explained by the reduction of ribosome biogenesis and protein translation, which can preserve more energy for cells to endure the damage induced by stroke.
Traumatic brain injury
Traumatic brain injury (TBI) is typically caused by an external mechanical force. The brain injuries caused by TBI can be divided into two categories: primary brain injury that occurs at the time of the insult, and secondary brain injury, which includes neuronal apoptosis, inflammation, oxidative stress, etc. [286]. Selective autophagy has been implicated in traumatic brain injury (TBI), as well.
Numerous studies have revealed that mitochondria might be critical for the pathophysiology of TBI. As the main site of energy production and oxidative metabolism, any disturbance of mitochondrial functions would lead to fuel deficiency, oxidative stress, and even induction of apoptosis, which is the key process of neural damages in TBI [287, 288]. Hence, mitophagy can alleviate secondary brain injuries by selective degradation of damaged mitochondria after TBI [289]. Wu et al. [290] have used mitochondrial division inhibitor-1 (Mdivi-1) to inhibit the key regulator of mitochondrial fission, Drp1, and have reported that it can extenuate TBI-induced blood–brain barrier (BBB) disruption and cell death by inhibiting dysfunctional autophagy, but by activating mitophagy. Likewise, Liu et al. [291] have reported increased mitophagy after TBI, which diminished the TBI-mediated intestinal epithelial cell damage, and improved intestinal permeability via ERK/Nrf2/HO-1 signaling. Mitophagy can negatively regulate IL-1β secretion, and thus inflammatory activation, to protect against TBI-triggered immunopathology [289]. The neuroprotective effects of mitophagy have also been demonstrated in spinal cord injury, which can be induced by inhibition of miRNA-124 or autophagy inducers, such as rapamycin [292,293,294].
Others
In addition to the aforementioned neurological diseases, selective autophagy has been implicated to play an important role in other neurological diseases as well, such as Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and infectious diseases of the CNS.
Huntington’s disease is an autosomal dominant neurodegenerative disease characterized by motor and cognitive impairment. The marked pathological features of HD include the formation of Huntingtin (Htt) aggregates and inclusions, which are mainly composed of Htt fragments with prolonged polyglutamine sequences (PolyQ). Some researchers have demonstrated that aggrephagy initiation greatly decreases the level of aggregated and soluble monomeric Htt species [295, 296]. Moreover, K63-ubiquitinated Htt has been shown to facilitate the target of aggregates by autophagy receptors, such as optineurin or p62 [297,298,299]. Besides, in preclinical HD models, growing studies have pointed to the role of mitochondrial Htt (mHtt) in mitochondrial functions and mitophagy [300]. Similarly, Valosin-containing protein (VCP), a mHtt-binding protein, is reportedly recruited to the mitochondria, leading to impaired mitophagy in models of HD [301].
Niemann–Pick disease type c is another rare, but fatal neurodegenerative disease, which is induced by genetic mutations in NPC1 (I1061T NPC1). The NPC1, a multipass transmembrane glycoprotein, is required for intracellular lipid delivery. Interestingly, Schultz et al. [302] suggested that I1061T NPC1 is selectively cleared by ER-phagy.
Amyotrophic lateral sclerosis is a neurodegenerative disease characterized by selective and progressive death of the motor neurons. Ubiquitinated inclusion bodies can be seen in the cytoplasm of these neuronal cells. Many studies have shown that mitochondrial dysfunction is a key factor in the pathogenesis of this disease. Studies have shown that mitophagy has a protective effect on ALS [303]. Specifically, dysfunction of mitophagy, due to ALS-associated mutants, is considered vital for causing mitochondrial dysfunction and accumulation, a prevalent feature in the motor neurons of ALS patients. Besides, aberrant mitochondrial axonal delivery is thought to be another factor contributing to the pathogenesis of ALS. Mitochondria from the soma are anterogradely transported to sites where the metabolism is vigorous, whereas abnormal functions of transportation lead to neuronal defects [304, 305]. Moreover, it is reported that autophagy was closely associated with DNA repair to promote neurodegeneration in ALS [306]. ALS can cause p62 mutations in or around the LC3 domain of p62, leading to autophagic defects and the accumulation of mutant p62, while the accumulation of p62 impairs the DNA damage response [307, 308]. Therefore, autophagy could be a promising target in treating ALS.
Mitochondrial dysfunction is also associated with infective processes [309], as the mitochondria have a number of roles in resisting bacterial infection, including the production of bactericidal ROS [310] and inflammasome activation [311]. Indeed, infections result in mitochondrial damage through an unknown mechanism, leading to the release of mitochondrial DNA (mtDNA) and mitochondrial ROS (mtROS) from the damaged mitochondria, which are thought to work as danger signals [312, 313]. As mentioned earlier, mitophagy is a regulatory mechanism of cells, which functions to eliminate damaged mitochondria to maintain mitochondrial homeostasis against stress and apoptosis [314, 315]. Among many others, HIV infection has been the most studied CNS infectious disease associated with mitophagy thus far. It is well known that HIV enters the CNS during the early stages of the infection, resulting in neurodegeneration and neurocognitive impairment. In HIV-productively infected astrocytes, mitophagy is crucial for cell death resistance. Moreover, mitophagy can reduce inflammation to counteract inflammasome activation; however, impaired mitophagy may favor inflammasome-mediated cell death in abortively infected cells [316]. In human primary neurons, HIV proteins, including gp120 and Tat, can cause neuronal degeneration, and thus, neurocognitive impairment by favoring the balance of mitochondrial dynamics toward enhanced fragmentation by activating mitochondrial translocation of DRP1 to the damaged mitochondria. Hence, a failure in completing the mitophagy process leads to neuronal damage [317].
From bench to bedside: neuroprotection of selective autophagy
Although neuronal autophagy is greatly decreased when compared with other tissues, the normal development and functions of the CNS are more dependent on basal autophagy than that of other tissues [318]. The cellular division in the CNS is mainly located at developmental stages, and mature neurons have a limited or null potential of proliferation, which indicates that damaged organelles and misfolded proteins cannot be redistributed and removed by division, and finally accumulate in the neurons, unless they are successfully removed by autophagy [319, 320]. However, the cytoprotective role of autophagy in neuronal tissues was firmly proved by establishing CNS-specific autophagy-deficient animal models [321]. The neural tissue-specific knockout models for essential autophagy genes show significant signs of neurodegeneration, including growth retardation, progressive motor deficits, abnormal reflexes, and often premature death [322,323,324].
Homeostatic and housekee** functions of autophagy in CNS
Different from other cell types, the normal functions of neuronal cells greatly depend on basal autophagy, as they are post-mitotic and suffer from aggregation of toxic proteins and damaged organelles over an extended period [325, 326]. Basal autophagy showed an important role in the regulation of axonal, dendritic, and synaptic homeostasis [327]. For example, Komatsu et al. [324] reported that the loss of basal autophagy by specific knockout of the autophagic gene, Atg7, in Purkinje cells resulted in progressive dystrophy and degeneration of the axon terminals of these cells. In addition, Lee et al. [328] demonstrated the role of mTOR in regulating post-synaptic potentiation or depression, which suggested that the effects of autophagy are involved in synaptic plasticity. Taken together, basal autophagy is relatively active in healthy neurons and maintains homeostasis via degradation of accumulated proteins and dysfunctional cell organelles.
Role of induced autophagy in neuroprotection
Growing evidence suggests that pharmacological induction of autophagic flux provides a promising clinical strategy for the treatment of neurological diseases. For example, rapamycin and its analogues, known as the ‘rapalogues’, reportedly enhance autophagosome formation by suppressing the functions of mTORC1, protecting against the toxicity of accumulated proteins in vitro, and substantially reducing neurodegeneration in fly and mouse models of HD and SCA3 [295, 329,330,331]. However, the beneficial effects of rapamycin treatment are greatly decreased in Drosophila models of HD and SCA3 when autophagy is inhibited [330, 332, 333]. Besides, virally delivered Beclin 1 reduced the neuropathology in mouse models of AD and Parkinson/Lewy body diseases [244, 334], which suggests that induction of autophagy enhances the neuroprotective effects of rapamycin. Consistently, other chemical agents capable of inducing autophagy in an mTOR-independent manner, such as carbamazepine, the molecular chaperone trehalose, the inositol monophosphatase inhibitors lithium, or valproate, increase the degradation of mutant huntingtin and protect against its toxicity in several models of neurodegeneration [335,336,337]. In addition, NAD+-induced mitophagy was also reported to reduce the cognitive loss of AD by enhancing the functions of sirtuins (SIRT1 to SIRT7), SARM1 (sterile alpha and TIR motif containing 1), and PARP (poly[ADP-ribose] polymerase) proteins [230]. Besides, decreased autophagy around the hematoma, exacerbation of neurological deficits, and brain edema in an intracerebral hemorrhage model with hyperglycemia indicate the beneficial role of autophagy in ICH with hyperglycemia [338]. For selective autophagy, the neuroprotective role of mitophagy has also been reported by many studies. Its underlying mechanisms include mitochondrial clearance and inhibition of downstream oxidative stress, apoptosis, and inflammation [339, 340]. One study showed that rapamycin could attenuate mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke [341]. In addition, several mitophagy-related proteins, such as beclin1 and Parkin, were all reported to be beneficial in the treatment of ischemic brain injury [342, 343]. Moreover, the neuroprotective effects of mitophagy have also been reported in hemorrhagic stroke by many studies [344, 345].
Potential clinical values of selective autophagy in neurological diseases
Some clinical trials have been set to explore the potential therapeutic effects of autophagy in human diseases. For example, hydroxychloroquine (HCQ) is reported to be a clinically approved autophagy inhibitor, and has been used in cancer clinical trials (NCT00813423, NCT01023737, et al.) [346]. Besides, there are also some clinical trials studying the clinical significance of mitophagy and pexophagy in human diseases (NCT02472340 and NCT03856866). However, clinical translational applications of these drugs remain in the early stages. Current limitations include difficulties in methodology and selective drug development. One of the most challenging aspects regarding the translation of autophagy is the difficulty in dynamically evaluating autophagy in vivo. This limitation is quite important, as it decides the diagnosis and monitors the efficiency of any autophagy-based intervention. At the experimental level, tandem macroautophagy reporter mRFP-GFP-LC3 or intraventricular delivery of adeno-associated viruses to the brain have been reported to be useful methods in monitoring autophagy [347]. However, autophagy reporters are not available for use in the clinical setting yet. Therefore, develo** methods for monitoring autophagy will also be important for the clinical translation of autophagy-based drugs.
Conclusions and perspectives
In this review, we comprehensively discussed the underlying mechanisms of selective autophagy and its roles in neurological diseases. Basically, selective autophagy may be responsible for the organelle turnover, and it turns out to be an energy-efficient, fast, and precise way to deal with unwanted materials. Physiological selective autophagy is triggered by various stresses to maintain cellular homeostasis. Until now, a number of studies have mainly focused on the mechanisms of selective autophagy; however, some types of autophagy (ribophagy, ER-phagy, pexophagy, etc.) are still far from being understood. For example, no specific adaptors or receptors for ribophagy have been identified. Likewise, the process by which LDs are recognized and transported to the lysosomes remains unknown.
In addition to the mechanisms behind selective autophagy, we discussed the crosstalk between selective autophagy and other cellular processes, as selective autophagy exhibits a close relationship with apoptosis, neuroinflammation, oxidative stress, etc. Besides, most of the current studies focus on the regulation of non-selective autophagy, whereas activation of general autophagy is far from understanding the whole lysosomal–autophagy system in the cells [348]. Therefore, achieving control of selective autophagy may be a promising strategy in preventing and treating neurological diseases. However, understanding the mechanisms of selective autophagy that are behind neurological diseases has been limited to preclinical animal studies with no clinical evidence reported thus far. Indeed, it is mandatory to determine how selective autophagy mechanisms occur in the human body. We need to understand the underlying mechanisms of selective autophagy, and the selective autophagy-connected crosstalk mechanisms. Broader selective reagents and therapeutic targets for the manipulation of selective autophagy are necessary. Finally, further elucidation of selective autophagy, as well as its crosstalk mechanisms under pathologic neurological conditions is warranted.
Abbreviations
- AD:
-
Alzheimer’s disease
- PD:
-
Parkinson’s disease
- PINK:
-
PTEN-induced kinase 1
- OPTN:
-
Optineurin
- NBR1:
-
Neighbor of BRCA1 gene 1
- NUFIP1:
-
Nuclear fragile X mental retardation-interacting protein 1
- NDP52:
-
Nuclear domain 10 protein 52
- PAS:
-
Phagophore assembly site
- ULK:
-
UNC51-like kinase
- PI3K III:
-
Class III phosphatidylinositol 3-kinase
- LAMP2A:
-
Lysosome-associated membrane protein 2
- hsc70:
-
Heat shock cognate 70
- Hsp70:
-
Heat shock protein 70
- HIP:
-
Hsc 70-interacting protein
- HOP:
-
Hsp70–Hsp90 Organizing Protein
- TAX1BP1:
-
Tax1-Binding Protein 1
- PEX5:
-
Peroxin 5
- BBB:
-
Blood–brain barrier
- DFCP1:
-
Double FYVE domain-containing protein
- WIPI1:
-
WD Repeat Domain, Phosphoinositide Interacting 1
- ARIH1:
-
Ariadne RBR E3 Ubiquitin Protein Ligase 1
- SIAH1:
-
Siah E3 Ubiquitin Protein Ligase 1
- MUL1:
-
Mitochondrial E3 Ubiquitin Protein Ligase 1
- OPA1:
-
Fission through optic atrophy 1
- DRP1:
-
Dynamin-related protein
- AMPK:
-
5′ AMP-activated protein kinase
- mTOR:
-
Mammalian target of rapamycin
- CMA:
-
Chaperone-mediated autophagy
- ROS:
-
Reactive oxidative species
- Nrf2:
-
Nuclear factor erythroid 2–related factor 2
- USP8:
-
Ubiquitin-Specific Peptidase 8
- LIR:
-
LC3-interacting regions
- UBA:
-
Ubiquitin-associated
- UBD:
-
Ubiquitin-binding domain
- CCPG-1:
-
Cell-cycle progression gene 1
- NIX:
-
NIP3-like protein X
- FAM134B:
-
Family with sequence similarity 134
- member B:
-
Bcl2L13, BCL2-like 13
- PGAM5:
-
Phosphoglycerate mutase 5
- TRIM50:
-
Tripartite Motif Containing 50
- BER:
-
Base excision repair
- DSB:
-
Double-strand breaks
- Bnip3:
-
BCL2/adenovirus E1B interacting protein 3
- AMBRA1:
-
Autophagy/Beclin 1 regulator 1
- FUNDC1:
-
FUN14 domain-containing 1
- PMI:
-
p62–SQSTM1-mediated mitophagy inducer
- DUB:
-
Deubiquitinating enzyme
- RNS:
-
Reactive nitrogen species
- VSM:
-
Vacuolar sequestering membrane
- MIPA:
-
Micropexophagy-specific apparatus
- ATM:
-
Ataxia-telangiectasia mutated
- RHD:
-
Reticulon homology domain
- GABARAP:
-
Gamma-aminobutyric acid receptor-associated protein
- GIM:
-
GABARAP-interacting motif
- LD:
-
Lipid droplet
- RTN3:
-
Reticulon 3
- CCPG1:
-
Cell-cycle progression 1
- ATL3:
-
Atlastin GTPase 3
- RTN3L:
-
Long isoform of RTN3
- HDAC6:
-
Histone deacetylase 6
- ALFY:
-
Autophagy-linked FYVE domain-containing protein
- ESCRT:
-
Endosomal sorting complex required for transport
- MPTP:
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- PARP-1:
-
Mitochondrial intermembrane space
- ALS:
-
Amyotrophic lateral sclerosis
- ERRα:
-
Estrogen-related receptor α
- GABA:
-
γ-Aminobutyric acid
- LDL:
-
Low-density lipid
- TBI:
-
Traumatic brain injury
- HD:
-
Huntington’s disease
- VCP:
-
Valosin-containing protein
References
Chrousos GP, Gold PW (1992) The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA 267(9):1244–1252
Corrigan JD, Selassie AW, Orman JA (2010) The epidemiology of traumatic brain injury. J Head Trauma Rehabil 25(2):72–80
Mayeux R (2003) Epidemiology of neurodegeneration. Annu Rev Neurosci 26:81–104
Bramlett HM, Dietrich WD (2004) Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab 24(2):133–150
Bossy-Wetzel E, Schwarzenbacher R, Lipton SA (2004) Molecular pathways to neurodegeneration. Nat Med 10(Suppl):S2–S9
Manczak M et al (2006) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15(9):1437–1449
Cutler RG et al (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101(7):2070–2075
Hanzel CE et al (2014) Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol Aging 35(10):2249–2262
Moore DJ et al (2005) Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci 28:57–87
Dawson TM, Dawson VL (2003) Molecular pathways of neurodegeneration in Parkinson’s disease. Science 302(5646):819–822
Yan F et al (2017) Pharmacological inhibition of PERK attenuates early brain injury after subarachnoid hemorrhage in rats through the activation of Akt. Mol Neurobiol 54(3):1808–1817
Xu W et al (2019) Sodium benzoate attenuates secondary brain injury by inhibiting neuronal apoptosis and reducing mitochondria-mediated oxidative stress in a rat model of intracerebral hemorrhage: possible involvement of DJ-1/Akt/IKK/NFkappaB pathway. Front Mol Neurosci 12:105
Clark SL Jr (1957) Cellular differentiation in the kidneys of newborn mice studies with the electron microscope. J Biophys Biochem Cytol 3(3):349–362
Deter RL, De Duve C (1967) Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol 33(2):437–449
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12
Bar-Yosef T, Damri O, Agam G (2019) Dual role of autophagy in diseases of the central nervous system. Front Cell Neurosci 13:196
Johansen T, Lamark T (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7(3):279–296
Yorimitsu T, Klionsky DJ (2005) Autophagy: molecular machinery for self-eating. Cell Death Differ 12(Suppl 2):1542–1552
Moloudizargari M et al (2017) Autophagy, its mechanisms and regulation: implications in neurodegenerative diseases. Ageing Res Rev 40:64–74
Lamb CA, Yoshimori T, Tooze SA (2013) The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol 14(12):759–774
Menon MB, Dhamija S (2018) Beclin 1 phosphorylation—at the center of autophagy regulation. Front Cell Dev Biol 6:137
Wang P et al (2018) Autophagy in ischemic stroke. Prog Neurobiol 163–164:98–117
Galluzzi L et al (2017) Molecular definitions of autophagy and related processes. EMBO J 36(13):1811–1836
Sahu R et al (2011) Microautophagy of cytosolic proteins by late endosomes. Dev Cell 20(1):131–139
Kaushik S, Cuervo AM (2012) Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22(8):407–417
Schneider JL, Cuervo AM (2014) Liver autophagy: much more than just taking out the trash. Nat Rev Gastroenterol Hepatol 11(3):187–200
Bandyopadhyay U et al (2008) The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol 28(18):5747–5763
Johansen T (2020) Lamark T (2019) Selective autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J Mol Biol 432(1):80–103
Lou G et al (2020) Mitophagy and neuroprotection. Trends Mol Med 26(1):8–20
Wang Y, Liu N, Lu B (2019) Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci Ther 25(7):859–875
Manjithaya R et al (2010) Molecular mechanism and physiological role of pexophagy. FEBS Lett 584(7):1367–1373
Wang Y et al (2019) Dexmedetomidine alleviates LPS-induced apoptosis and inflammation in macrophages by eliminating damaged mitochondria via PINK1 mediated mitophagy. Int Immunopharmacol 73:471–481
Vasko R et al (2013) Endothelial peroxisomal dysfunction and impaired pexophagy promotes oxidative damage in lipopolysaccharide-induced acute kidney injury. Antioxid Redox Signal 19(3):211–230
Azodi S, Jacobson S (2016) Cytokine therapies in neurological disease. Neurotherapeutics 13(3):555–561
Longo FM, Massa SM (2013) Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov 12(7):507–525
Lejri I et al (2019) Mitochondria- and oxidative stress-targeting substances in cognitive decline-related disorders: from molecular mechanisms to clinical evidence. Oxid Med Cell Longev 2019:9695412
Chen Y, Zhou Z, Min W (2018) Mitochondria, oxidative stress and innate immunity. Front Physiol 9:1487
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8(1):3–5
Saito T, Sadoshima J (2015) Molecular mechanisms of mitochondrial autophagy/mitophagy in the heart. Circ Res 116(8):1477–1490
Hamacher-Brady A, Brady NR (2016) Mitophagy programs: mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell Mol Life Sci 73(4):775–795
Tan S, Wong E (2017) Mitophagy transcriptome: mechanistic insights into polyphenol-mediated mitophagy. Oxid Med Cell Longev 2017:9028435
Geisler S et al (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12(2):119–131
Matsuda N et al (2010) PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189(2):211–221
Narendra D et al (2008) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183(5):795–803
Vives-Bauza C et al (2010) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107(1):378–383
Chaugule VK et al (2011) Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J 30(14):2853–2867
Chu CT (2019) Mechanisms of selective autophagy and mitophagy: implications for neurodegenerative diseases. Neurobiol Dis 122:23–34
Yamaguchi O et al (2016) Receptor-mediated mitophagy. J Mol Cell Cardiol 95:50–56
Lazarou M et al (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524(7565):309–314
Yoo SM, Jung YK (2018) A molecular approach to mitophagy and mitochondrial dynamics. Mol Cells 41(1):18–26
Strappazzon F et al (2015) AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1. Cell Death Differ 22(3):419–432
McWilliams TG et al (2016) mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J Cell Biol 214(3):333–345
Sun N et al (2015) Measuring in vivo mitophagy. Mol Cell 60(4):685–696
Esteban-Martinez L et al (2017) Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36(12):1688–1706
Sandoval H et al (2008) Essential role for Nix in autophagic maturation of erythroid cells. Nature 454(7201):232–235
Schweers RL et al (2007) NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104(49):19500–19505
Sekine S, Youle RJ (2018) PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol 16(1):2
Pickles S, Vigie P, Youle RJ (2018) Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol 28(4):R170–R185
Lazarou M et al (2012) Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22(2):320–333
Deas E et al (2011) PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20(5):867–879
Greene AW et al (2012) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13(4):378–385
** SM et al (2010) Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191(5):933–942
Meissner C et al (2011) The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 117(5):856–867
Yamano K, Youle RJ (2013) PINK1 is degraded through the N-end rule pathway. Autophagy 9(11):1758–1769
Harper JW, Ordureau A, Heo JM (2018) Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol 19(2):93–108
Hasson SA et al (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504(7479):291–295
Kondapalli C et al (2012) PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2(5):120080
Okatsu K et al (2012) PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3:1016
Aerts L et al (2015) PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290(5):2798–2811
Okatsu K et al (2013) A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288(51):36372–36384
Matsuda N (2016) Phospho-ubiquitin: upending the PINK–Parkin–ubiquitin cascade. J Biochem 159(4):379–385
Shiba-Fukushima K et al (2012) PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2:1002
Kane LA et al (2014) PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205(2):143–153
Koyano F et al (2014) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510(7503):162–166
Kazlauskaite A et al (2014) Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460(1):127–139
Nardin A, Schrepfer E, Ziviani E (2016) Counteracting PINK/Parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson’s disease. Curr Neuropharmacol 14(3):250–259
Aguirre JD et al (2017) Structure of phosphorylated UBL domain and insights into PINK1-orchestrated parkin activation. Proc Natl Acad Sci USA 114(2):298–303
Yu H et al (2019) Association of the TBK1 mutation p.Ile334Thr with frontotemporal dementia and literature review. Mol Genet Genomic Med 7(3):e547
Abudu YP et al (2019) NIPSNAP1 and NIPSNAP2 act as “eat me” signals to allow sustained recruitment of autophagy receptors during mitophagy. Autophagy 15(10):1845–1847
Fu M et al (2013) Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 24(8):1153–1162
Lokireddy S et al (2012) The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab 16(5):613–624
Orvedahl A et al (2011) Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480(7375):113–117
Szargel R et al (2016) The PINK1, synphilin-1 and SIAH-1 complex constitutes a novel mitophagy pathway. Hum Mol Genet 25(16):3476–3490
Villa E et al (2017) Parkin-independent mitophagy controls chemotherapeutic response in cancer cells. Cell Rep 20(12):2846–2859
Gatica D, Lahiri V, Klionsky DJ (2018) Cargo recognition and degradation by selective autophagy. Nat Cell Biol 20(3):233–242
Murakawa T et al (2015) Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6:7527
Diwan A et al (2007) Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Investig 117(10):2825–2833
Schwarten M et al (2009) Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5(5):690–698
Rogov VV et al (2017) Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep 7(1):1131
Melser S et al (2013) Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 17(5):719–730
Quinsay MN et al (2010) Bnip3 mediates permeabilization of mitochondria and release of cytochrome c via a novel mechanism. J Mol Cell Cardiol 48(6):1146–1156
Quinsay MN et al (2010) Bnip3-mediated mitochondrial autophagy is independent of the mitochondrial permeability transition pore. Autophagy 6(7):855–862
Zhang T et al (2016) BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol Chem 291(41):21616–21629
Lee Y et al (2011) Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am J Physiol Heart Circ Physiol 301(5):H1924–H1931
Liu L et al (2012) Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14(2):177–185
Wu W et al (2016) FUNDC1 regulates mitochondrial dynamics at the ER-mitochondrial contact site under hypoxic conditions. EMBO J 35(13):1368–1384
Palikaras K, Lionaki E, Tavernarakis N (2018) Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol 20(9):1013–1022
Shi RY et al (2014) BNIP3 interacting with LC3 triggers excessive mitophagy in delayed neuronal death in stroke. CNS Neurosci Ther 20(12):1045–1055
Chakrabarti L et al (2009) Autophagy activation and enhanced mitophagy characterize the Purkinje cells of pcd mice prior to neuronal death. Mol Brain 2:24
Galluzzi L et al (2014) Metabolic control of autophagy. Cell 159(6):1263–1276
Hertz NT et al (2013) A neo-substrate that amplifies catalytic activity of Parkinson’s-disease-related kinase PINK1. Cell 154(4):737–747
Hasson SA et al (2015) Chemogenomic profiling of endogenous PARK2 expression using a genome-edited coincidence reporter. ACS Chem Biol 10(5):1188–1197
Komander D (2010) Mechanism, specificity and structure of the deubiquitinases. Subcell Biochem 54:69–87
Cornelissen T et al (2014) The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23(19):5227–5242
Faronato M et al (2013) The deubiquitylase USP15 stabilizes newly synthesized REST and rescues its expression at mitotic exit. Cell Cycle 12(12):1964–1977
Eichhorn PJ et al (2012) USP15 stabilizes TGF-beta receptor I and promotes oncogenesis through the activation of TGF-beta signaling in glioblastoma. Nat Med 18(3):429–435
Herhaus L et al (2014) USP15 targets ALK3/BMPR1A for deubiquitylation to enhance bone morphogenetic protein signalling. Open Biol 4(5):140065
Inui M et al (2011) USP15 is a deubiquitylating enzyme for receptor-activated SMADs. Nat Cell Biol 13(11):1368–1375
Zhang H et al (2015) Ubiquitin-specific protease 15 negatively regulates virus-induced type I interferon signaling via catalytically-dependent and -independent mechanisms. Sci Rep 5:11220
Zou Q et al (2014) USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol 15(6):562–570
Wang Y et al (2015) Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11(4):595–606
MacDonald E, Urbe S, Clague MJ (2014) USP8 controls the trafficking and sorting of lysosomal enzymes. Traffic 15(8):879–888
Durcan TM et al (2014) USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33(21):2473–2491
Eiyama A, Okamoto K (2015) PINK1/Parkin-mediated mitophagy in mammalian cells. Curr Opin Cell Biol 33:95–101
Durcan TM, Fon EA (2015) The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev 29(10):989–999
Wang L et al (2018) PTEN-L is a novel protein phosphatase for ubiquitin dephosphorylation to inhibit PINK1-Parkin-mediated mitophagy. Cell Res 28(8):787–802
Taylor R, Goldman SJ (2011) Mitophagy and disease: new avenues for pharmacological intervention. Curr Pharm Des 17(20):2056–2073
Wang L et al (2020) Post-translational modifications of key machinery in the control of mitophagy. Trends Biochem Sci 45(1):58–75
Faust PL, Kovacs WJ (2014) Cholesterol biosynthesis and ER stress in peroxisome deficiency. Biochimie 98:75–85
Fransen M et al (2012) Role of peroxisomes in ROS/RNS-metabolism: implications for human disease. Biochim Biophys Acta 1822(9):1363–1373
Schonenberger MJ, Kovacs WJ (2015) Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol 3:42
Van Veldhoven PP (2010) Biochemistry and genetics of inherited disorders of peroxisomal fatty acid metabolism. J Lipid Res 51(10):2863–2895
Walker CL et al (2018) Redox regulation of homeostasis and proteostasis in peroxisomes. Physiol Rev 98(1):89–115
Scott SV, Klionsky DJ (1998) Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr Opin Cell Biol 10(4):523–529
Sakai Y et al (1998) Peroxisome degradation by microautophagy in Pichia pastoris: identification of specific steps and morphological intermediates. J Cell Biol 141(3):625–636
Veenhuis M et al (1983) Degradation and turnover of peroxisomes in the yeast Hansenula polymorpha induced by selective inactivation of peroxisomal enzymes. Arch Microbiol 134(3):193–203
Tuttle DL, Dunn WA Jr (1995) Divergent modes of autophagy in the methylotrophic yeast Pichia pastoris. J Cell Sci 108(Pt 1):25–35
Mukaiyama H et al (2004) Modification of a ubiquitin-like protein Paz2 conducted micropexophagy through formation of a novel membrane structure. Mol Biol Cell 15(1):58–70
Farre JC et al (2009) Turnover of organelles by autophagy in yeast. Curr Opin Cell Biol 21(4):522–530
Mancias JD, Kimmelman AC (2016) Mechanisms of selective autophagy in normal physiology and cancer. J Mol Biol 428(9 Pt A):1659–1680
Kirkin V et al (2009) A role for ubiquitin in selective autophagy. Mol Cell 34(3):259–269
Deosaran E et al (2013) NBR1 acts as an autophagy receptor for peroxisomes. J Cell Sci 126(Pt 4):939–952
Anding AL, Baehrecke EH (2017) Cleaning house: selective autophagy of organelles. Dev Cell 41(1):10–22
Katsuragi Y, Ichimura Y, Komatsu M (2015) p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282(24):4672–4678
Ryter SW, Cloonan SM, Choi AM (2013) Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells 36(1):7–16
Hara-Kuge S, Fujiki Y (2008) The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp Cell Res 314(19):3531–3541
Jiang L et al (2015) Peroxin Pex14p is the key component for coordinated autophagic degradation of mammalian peroxisomes by direct binding to LC3-II. Genes Cells 20(1):36–49
Zhang J et al (2015) ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat Cell Biol 17(10):1259–1269
Alexander A et al (2010) ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA 107(9):4153–4158
Feng L et al (2017) Ubiquitin ligase SYVN1/HRD1 facilitates degradation of the SERPINA1 Z variant/alpha-1-antitrypsin Z variant via SQSTM1/p62-dependent selective autophagy. Autophagy 13(4):686–702
Yamashita S et al (2014) The membrane peroxin PEX3 induces peroxisome-ubiquitination-linked pexophagy. Autophagy 10(9):1549–1564
Vadlamudi RK et al (1996) p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem 271(34):20235–20237
Bjorkoy G et al (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171(4):603–614
Komatsu M et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131(6):1149–1163
Ichimura Y et al (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 283(33):22847–22857
Kirkin V et al (2009) A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 33(4):505–516
Mardakheh FK et al (2010) Nbr1 is a novel inhibitor of ligand-mediated receptor tyrosine kinase degradation. Mol Cell Biol 30(24):5672–5685
Zientara-Rytter K, Subramani S (2016) Autophagic degradation of peroxisomes in mammals. Biochem Soc Trans 44(2):431–440
Zhang B et al (2014) Involvement of the Nrf2 pathway in the regulation of pterostilbene-induced apoptosis in HeLa cells via ER stress. J Pharmacol Sci 126(3):216–229
Zhang S et al (2017) The lectin chaperone calnexin is involved in the endoplasmic reticulum stress response by regulating Ca(2+) homeostasis in Aspergillus nidulans. Appl Environ Microbiol 83(15):e00673–17
Zhong Q et al (2011) Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: effects of misfolded surfactant protein. Am J Respir Cell Mol Biol 45(3):498–509
Bernales S, Schuck S, Walter P (2007) ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3(3):285–287
Khaminets A et al (2015) Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522(7556):354–358
Voeltz GK et al (2006) A class of membrane proteins sha** the tubular endoplasmic reticulum. Cell 124(3):573–586
Grumati P et al (2017) Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. eLife 6:e25555
Edwards MC et al (1997) Human CPR (cell cycle progression restoration) genes impart a Far− phenotype on yeast cells. Genetics 147(3):1063–1076
Smith MD et al (2018) CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev Cell 44(2):217–232e11
Chen Q, Teng J, Chen J (2019) ATL3, a cargo receptor for reticulophagy. Autophagy 15(8):1465–1466
Mizushima N, Ohsumi Y, Yoshimori T (2002) Autophagosome formation in mammalian cells. Cell Struct Funct 27(6):421–429
Chen Q et al (2019) ATL3 is a tubular ER-phagy receptor for GABARAP-mediated selective autophagy. Curr Biol 29(5):846–855e6
Forrester A et al (2019) A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38(2):e99847
Chiramel AI et al (2016) FAM134B, the selective autophagy receptor for endoplasmic reticulum turnover, inhibits replication of Ebola virus strains Makona and Mayinga. J Infect Dis 214(suppl 3):S319–S325
Wilkinson S (2019) ER-phagy: sha** up and destressing the endoplasmic reticulum. FEBS J 286(14):2645–2663
Chung KK, Dawson VL, Dawson TM (2001) The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci 24(11 Suppl):S7–S14
Kaarniranta K et al (2009) Heat shock proteins as gatekeepers of proteolytic pathways—implications for age-related macular degeneration (AMD). Ageing Res Rev 8(2):128–139
Wang AL et al (2009) Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One 4(1):e4160
Shiarli AM et al (2006) Comparison of extent of tau pathology in patients with frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17), frontotemporal lobar degeneration with Pick bodies and early onset Alzheimer’s disease. Neuropathol Appl Neurobiol 32(4):374–387
Sun D et al (2020) Phase separation in regulation of aggrephagy. J Mol Biol 432(1):160–169
Hyttinen JM et al (2014) Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev 18:16–28
Viiri J et al (2010) p62/sequestosome 1 as a regulator of proteasome inhibitor-induced autophagy in human retinal pigment epithelial cells. Mol Vis 16:1399–1414
Jung H et al (2020) NBR1 mediates selective autophagy of defective proteins in Arabidopsis. J Exp Bot 71(1):73–89
Su M et al (2011) HDAC6 regulates aggresome-autophagy degradation pathway of alpha-synuclein in response to MPP+-induced stress. J Neurochem 117(1):112–120
Filimonenko M et al (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell 38(2):265–279
Lamark T, Johansen T (2012) Aggrephagy: selective disposal of protein aggregates by macroautophagy. Int J Cell Biol 2012:736905
Shi CS, Kehrl JH (2010) TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci Signal 3(123):ra42
Wooten MW et al (2005) The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem 280(42):35625–35629
Lystad AH et al (2014) Structural determinants in GABARAP required for the selective binding and recruitment of ALFY to LC3B-positive structures. EMBO Rep 15(5):557–565
Isakson P, Holland P, Simonsen A (2013) The role of ALFY in selective autophagy. Cell Death Differ 20(1):12–20
Fusco C et al (2012) The E3-ubiquitin ligase TRIM50 interacts with HDAC6 and p62, and promotes the sequestration and clearance of ubiquitinated proteins into the aggresome. PLoS One 7(7):e40440
Tammineni P et al (2017) Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 6:e21776
Iwata A et al (2005) HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem 280(48):40282–40292
Li Y, Shin D, Kwon SH (2013) Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J 280(3):775–793
Kawaguchi Y et al (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115(6):727–738
Kraft C et al (2008) Mature ribosomes are selectively degraded upon starvation by an autophagy pathway requiring the Ubp3p/Bre5p ubiquitin protease. Nat Cell Biol 10(5):602–610
Ossareh-Nazari B et al (2014) Ubiquitylation by the Ltn1 E3 ligase protects 60S ribosomes from starvation-induced selective autophagy. J Cell Biol 204(6):909–917
Wyant GA et al (2018) NUFIP1 is a ribosome receptor for starvation-induced ribophagy. Science 360(6390):751–758
Schulze RJ, Sathyanarayan A, Mashek DG (2017) Breaking fat: the regulation and mechanisms of lipophagy. Biochim Biophys Acta Mol Cell Biol Lipids 1862(10 Pt B):1178–1187
Wang CW (2016) Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta 1861(8 Pt B):793–805
Weidberg H, Shvets E, Elazar Z (2009) Lipophagy: selective catabolism designed for lipids. Dev Cell 16(5):628–630
Singh R, Cuervo AM (2012) Lipophagy: connecting autophagy and lipid metabolism. Int J Cell Biol 2012:282041
Singh R et al (2009) Autophagy regulates lipid metabolism. Nature 458(7242):1131–1135
Martinez-Lopez N, Singh R (2016) Telemetric control of peripheral lipophagy by hypothalamic autophagy. Autophagy 12(8):1404–1405
Gomez-Sintes R, Ledesma MD, Boya P (2016) Lysosomal cell death mechanisms in aging. Ageing Res Rev 32:150–168
Radulovic M et al (2018) ESCRT-mediated lysosome repair precedes lysophagy and promotes cell survival. EMBO J 37(21):e99753
Papadopoulos C, Kravic B, Meyer H (2020) Repair or lysophagy: dealing with damaged lysosomes. J Mol Biol 432(1):231–239
Chu YP et al (2017) Assays to monitor lysophagy. Methods Enzymol 588:231–244
Ravenhill BJ et al (2019) The cargo receptor NDP52 initiates selective autophagy by recruiting the ULK complex to cytosol-invading bacteria. Mol Cell 74(2):320–329e6
Koerver L et al (2019) The ubiquitin-conjugating enzyme UBE2QL1 coordinates lysophagy in response to endolysosomal damage. EMBO Rep 20(10):e48014
Fayyad M et al (2019) Parkinson’s disease biomarkers based on alpha-synuclein. J Neurochem 150(5):626–636
Wakabayashi K et al (2007) The Lewy body in Parkinson’s disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 27(5):494–506
Lykkebo S, Jensen PH (2002) Alpha-synuclein and presynaptic function: implications for Parkinson’s disease. Neuromol Med 2(2):115–129
Luk KC et al (2012) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338(6109):949–953
Cheng HC, Ulane CM, Burke RE (2010) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67(6):715–725
Sn S et al (2019) Small molecule modulator of aggrephagy regulates neuroinflammation to curb pathogenesis of neurodegeneration. EBioMedicine 50:260–273
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997
Dikic I, Elazar Z (2018) Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol 19(6):349–364
Fujikake N, Shin M, Shimizu S (2018) Association between autophagy and neurodegenerative diseases. Front Neurosci 12:255
Wu MY et al (2018) Selective autophagy: the new player in the fight against neurodegenerative diseases? Brain Res Bull 137:79–90
Suresh SN et al (2018) Modulation of autophagy by a small molecule inverse agonist of ERRalpha is neuroprotective. Front Mol Neurosci 11:109
Schapira AH (2008) Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 7(1):97–109
Langston JW et al (1983) Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219(4587):979–980
Betarbet R et al (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3(12):1301–1306
Soutar MPM et al (2019) FBS/BSA media concentration determines CCCP’s ability to depolarize mitochondria and activate PINK1-PRKN mitophagy. Autophagy 15(11):1–10
Lee J, Giordano S, Zhang J (2012) Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J 441(2):523–540
Oh CK et al (2017) S-Nitrosylation of PINK1 attenuates PINK1/Parkin-dependent mitophagy in hiPSC-based Parkinson’s disease models. Cell Rep 21(8):2171–2182
Fang EF et al (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17(5):308–321
Tripathi DN et al (2013) Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proc Natl Acad Sci USA 110(32):E2950–E2957
Moreira PI et al (2010) Autophagy in Alzheimer’s disease. Expert Rev Neurother 10(7):1209–1218
Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362(4):329–344
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8(6):595–608
Weller RO, Boche D, Nicoll JA (2009) Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol 118(1):87–102
Moreira PI et al (2007) Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 66(6):525–532
Correia SC et al (2012) Alzheimer disease as a vascular disorder: where do mitochondria fit? Exp Gerontol 47(11):878–886
Viana RJ, Nunes AF, Rodrigues CM (2012) Endoplasmic reticulum enrollment in Alzheimer’s disease. Mol Neurobiol 46(2):522–534
Ma LY et al (2017) Autophagy-lysosome dysfunction is involved in Abeta deposition in STZ-induced diabetic rats. Behav Brain Res 320:484–493
Ling D, Magallanes M, Salvaterra PM (2014) Accumulation of amyloid-like Abeta1-42 in AEL (autophagy-endosomal-lysosomal) vesicles: potential implications for plaque biogenesis. ASN Neuro 6(2):e00139
Nilsson P, Saido TC (2014) Dual roles for autophagy: degradation and secretion of Alzheimer’s disease Abeta peptide. BioEssays 36(6):570–578
Nixon RA, Yang DS (2011) Autophagy failure in Alzheimer’s disease—locating the primary defect. Neurobiol Dis 43(1):38–45
Fang EF (2019) Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy 15(6):1112–1114
Fang EF et al (2019) Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci 22(3):401–412
Kerr JS et al (2017) Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci 40(3):151–166
Flannery PJ, Trushina E (2019) Mitochondrial dynamics and transport in Alzheimer’s disease. Mol Cell Neurosci 98:109–120
Martin-Maestro P et al (2017) Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s disease-associated presenilin 1 mutation. Front Mol Neurosci 10:291
Esposito L et al (2006) Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26(19):5167–5179
Mattson MP, Robinson N, Guo Q (1997) Estrogens stabilize mitochondrial function and protect neural cells against the pro-apoptotic action of mutant presenilin-1. NeuroReport 8(17):3817–3821
Hoglinger GU et al (2005) The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J Neurochem 95(4):930–939
Kondadi AK et al (2014) Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation. EMBO J 33(9):1011–1026
Fivenson EM et al (2017) Mitophagy in neurodegeneration and aging. Neurochem Int 109:202–209
Yang B et al (2019) NEIL1 stimulates neurogenesis and suppresses neuroinflammation after stress. Free Radic Biol Med 141:47–58
Hou Y et al (2019) Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 15(10):565–581
Sliwinska A et al (2017) Decreased expression level of BER genes in Alzheimer’s disease patients is not derivative of their DNA methylation status. Prog Neuropsychopharmacol Biol Psychiatry 79(Pt B):311–316
Shanbhag NM et al (2019) Early neuronal accumulation of DNA double strand breaks in Alzheimer’s disease. Acta Neuropathol Commun 7(1):77
Bai J, Liu F (2019) The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes 68(6):1099–1108
Pickford F et al (2008) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Investig 118(6):2190–2199
Rohn TT et al (2011) Depletion of Beclin-1 due to proteolytic cleavage by caspases in the Alzheimer’s disease brain. Neurobiol Dis 43(1):68–78
Lee JH et al (2010) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141(7):1146–1158
Babu JR, Geetha T, Wooten MW (2005) Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem 94(1):192–203
Kuusisto E, Salminen A, Alafuzoff I (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. NeuroReport 12(10):2085–2090
Caccamo A et al (2017) p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry 22(6):865–873
Decuypere JP et al (2011) The IP(3) receptor-mitochondria connection in apoptosis and autophagy. Biochim Biophys Acta 1813(5):1003–1013
Deegan S et al (2013) Stress-induced self-cannibalism: on the regulation of autophagy by endoplasmic reticulum stress. Cell Mol Life Sci 70(14):2425–2441
Schuck S, Gallagher CM, Walter P (2014) ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 127(Pt 18):4078–4088
Hoozemans JJ et al (2009) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174(4):1241–1251
Hoozemans JJ et al (2012) Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis 10(1–4):212–215
Lai CS et al (2009) Low molecular weight Abeta induces collapse of endoplasmic reticulum. Mol Cell Neurosci 41(1):32–43
Shibuya Y et al (2014) Inhibiting ACAT1/SOAT1 in microglia stimulates autophagy-mediated lysosomal proteolysis and increases Abeta1-42 clearance. J Neurosci 34(43):14484–14501
Zhou F et al (2011) APP and APLP1 are degraded through autophagy in response to proteasome inhibition in neuronal cells. Protein Cell 2(5):377–383
Loewen CA, Feany MB (2010) The unfolded protein response protects from tau neurotoxicity in vivo. PLoS One 5(9):e13084
Fester L et al (2009) Cholesterol-promoted synaptogenesis requires the conversion of cholesterol to estradiol in the hippocampus. Hippocampus 19(8):692–705
Goritz C, Mauch DH, Pfrieger FW (2005) Multiple mechanisms mediate cholesterol-induced synaptogenesis in a CNS neuron. Mol Cell Neurosci 29(2):190–201
de Chaves EI et al (1997) Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J Biol Chem 272(49):30766–30773
Liu Q et al (2010) Neuronal LRP1 knockout in adult mice leads to impaired brain lipid metabolism and progressive, age-dependent synapse loss and neurodegeneration. J Neurosci 30(50):17068–17078
Liu Q et al (2007) Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron 56(1):66–78
Linetti A et al (2010) Cholesterol reduction impairs exocytosis of synaptic vesicles. J Cell Sci 123(Pt 4):595–605
Jana A, Pahan K (2010) Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J Neurosci 30(38):12676–12689
Sanchez-Mejia RO et al (2008) Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat Neurosci 11(11):1311–1318
Esposito G et al (2008) Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med 49(9):1414–1421
Jung WH et al (2017) Lipophagy prevents activity-dependent neurodegeneration due to dihydroceramide accumulation in vivo. EMBO Rep 18(7):1150–1165
Wannamaker R, Buck B, Butcher K (2019) Multimodal CT in acute stroke. Curr Neurol Neurosci Rep 19(9):63
Guan R et al (2018) Mitophagy, a potential therapeutic target for stroke. J Biomed Sci 25(1):87
Jang I et al (2014) Genetic ablation and short-duration inhibition of lipoxygenase results in increased macroautophagy. Exp Cell Res 321(2):276–287
Lakhani R et al (2014) Defects in GABA metabolism affect selective autophagy pathways and are alleviated by mTOR inhibition. EMBO Mol Med 6(4):551–566
Zhu W et al (2017) Pexophagy is neuroprotective in ischemic brain. Stroke 48
Lee JS et al (2017) Triglyceride and HDL-C dyslipidemia and risks of coronary heart disease and ischemic stroke by glycemic dysregulation status: the strong heart study. Diabetes Care 40(4):529–537
Glasser SP et al (2016) What is the association of lipid levels and incident stroke? Int J Cardiol 220:890–894
Bostrom P et al (2006) Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol 26(8):1871–1876
Lam T et al (2016) Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov 2:16061
Unger RH, Orci L (2002) Lipoapoptosis: its mechanism and its diseases. Biochim Biophys Acta 1585(2–3):202–212
Ayala A, Munoz MF, Arguelles S (2014) Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014:360438
Cojocaru IM et al (2013) Evaluation of oxidative stress in patients with acute ischemic stroke. Rom J Intern Med 51(2):97–106
Kirac E et al (2015) Analysis of polyunsaturated fatty acids and the omega-6 inflammatory pathway in hepatic ischemia/re-perfusion injury. Mol Med Rep 12(3):4149–4156
Lonati E et al (2019) Lipid resha** and lipophagy are induced in a modeled ischemia-reperfusion injury of blood brain barrier. Int J Mol Sci 20(15):346
Kounakis K et al (2019) Emerging roles of lipophagy in health and disease. Front Cell Dev Biol 7:185
Bernales S, McDonald KL, Walter P (2006) Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4(12):e423
Carloni S et al (2014) Increased autophagy reduces endoplasmic reticulum stress after neonatal hypoxia–ischemia: role of protein synthesis and autophagic pathways. Exp Neurol 255:103–112
Hawryluk GW, Bullock MR (2016) Past, present, and future of traumatic brain injury research. Neurosurg Clin N Am 27(4):375–396
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443(7113):787–795
Gajavelli S et al (2015) Evidence to support mitochondrial neuroprotection, in severe traumatic brain injury. J Bioenerg Biomembr 47(1–2):133–148
Lin C et al (2016) Melatonin attenuates traumatic brain injury-induced inflammation: a possible role for mitophagy. J Pineal Res 61(2):177–186
Wu Q et al (2018) Mdivi-1 alleviates blood-brain barrier disruption and cell death in experimental traumatic brain injury by mitigating autophagy dysfunction and mitophagy activation. Int J Biochem Cell Biol 94:44–55
Liu Y et al (2017) Extracellular signal-regulated kinase/nuclear factor-erythroid2-like2/heme oxygenase-1 pathway-mediated mitophagy alleviates traumatic brain injury-induced intestinal mucosa damage and epithelial barrier dysfunction. J Neurotrauma 34(13):2119–2131
Liu K et al (2017) Acquired inhibition of microRNA-124 protects against spinal cord ischemia-reperfusion injury partially through a mitophagy-dependent pathway. J Thorac Cardiovasc Surg 154(5):1498–1508
Balsam LB (2017) Spinal cord ischemia-reperfusion injury: microRNAs and mitophagy at a crossroads. J Thorac Cardiovasc Surg 154(5):1509–1510
Li Q et al (2018) Rapamycin enhances mitophagy and attenuates apoptosis after spinal ischemia-reperfusion injury. Front Neurosci 12:865
Ravikumar B et al (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 36(6):585–595
Roscic A et al (2011) Induction of autophagy with catalytic mTOR inhibitors reduces huntingtin aggregates in a neuronal cell model. J Neurochem 119(2):398–407
Tan JM et al (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet 17(3):431–439
Korac J et al (2013) Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 126(Pt 2):580–592
Butland SL et al (2014) The palmitoyl acyltransferase HIP14 shares a high proportion of interactors with huntingtin: implications for a role in the pathogenesis of Huntington’s disease. Hum Mol Genet 23(15):4142–4160
Khalil B et al (2015) PINK1-induced mitophagy promotes neuroprotection in Huntington’s disease. Cell Death Dis 6:e1617
Guo X et al (2016) VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat Commun 7:12646
Schultz ML et al (2018) Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun 9(1):3671
Evans CS, Holzbaur ELF (2019) Autophagy and mitophagy in ALS. Neurobiol Dis 122:35–40
Hollenbeck PJ, Saxton WM (2005) The axonal transport of mitochondria. J Cell Sci 118(Pt 23):5411–5419
Kang JS et al (2008) Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132(1):137–148
Walker C, El-Khamisy SF (2018) Perturbed autophagy and DNA repair converge to promote neurodegeneration in amyotrophic lateral sclerosis and dementia. Brain 141(5):1247–1262
Rubino E et al (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 79(15):1556–1562
Goode A et al (2016) Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 12(7):1094–1104
Pellegrino MW, Haynes CM (2015) Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol 13:22
Arsenijevic D et al (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26(4):435–439
Zhou R et al (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469(7329):221–225
Nakahira K et al (2011) Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12(3):222–230
Shimada K et al (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36(3):401–414
Kim MJ, Yoon JH, Ryu JH (2016) Mitophagy: a balance regulator of NLRP3 inflammasome activation. BMB Rep 49(10):529–535
Zhu L et al (2016) Mitophagy in TGEV infection counteracts oxidative stress and apoptosis. Oncotarget 7(19):27122–27141
Ojeda DS et al (2018) Cell death is counteracted by mitophagy in HIV-productively infected astrocytes but is promoted by inflammasome activation among non-productively infected cells. Front Immunol 9:2633
Teodorof-Diedrich C, Spector SA (2018) Human immunodeficiency virus type 1 gp120 and Tat induce mitochondrial fragmentation and incomplete mitophagy in human neurons. J Virol 92(22):e00993–18
Mizushima N et al (2008) Autophagy fights disease through cellular self-digestion. Nature 451(7182):1069–1075
Winslow AR, Rubinsztein DC (2008) Autophagy in neurodegeneration and development. Biochim Biophys Acta 1782(12):723–729
Nixon RA, Yang DS, Lee JH (2008) Neurodegenerative lysosomal disorders: a continuum from development to late age. Autophagy 4(5):590–599
Nishiyama J et al (2007) Aberrant membranes and double-membrane structures accumulate in the axons of Atg5-null Purkinje cells before neuronal death. Autophagy 3(6):591–596
Hara T et al (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889
Komatsu M et al (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884
Komatsu M et al (2007) Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA 104(36):14489–14494
Tooze SA, Schiavo G (2008) Liaisons dangereuses: autophagy, neuronal survival and neurodegeneration. Curr Opin Neurobiol 18(5):504–515
Son JH et al (2012) Neuronal autophagy and neurodegenerative diseases. Exp Mol Med 44(2):89–98
Marino G, Madeo F, Kroemer G (2011) Autophagy for tissue homeostasis and neuroprotection. Curr Opin Cell Biol 23(2):198–206
Lee JA (2012) Neuronal autophagy: a housekeeper or a fighter in neuronal cell survival? Exp Neurobiol 21(1):1–8
Ravikumar B, Duden R, Rubinsztein DC (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum Mol Genet 11(9):1107–1117
Berger Z et al (2006) Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet 15(3):433–442
Menzies FM et al (2010) Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain 133(Pt 1):93–104
Pandey UB et al (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447(7146):859–863
Wang T, Lao U, Edgar BA (2009) TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol 186(5):703–711
Spencer B et al (2009) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29(43):13578–13588
Sarkar S et al (2008) A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum Mol Genet 17(2):170–178
Sarkar S et al (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170(7):1101–1111
Williams A et al (2008) Novel targets for Huntington’s disease in an mTOR-independent autophagy pathway. Nat Chem Biol 4(5):295–305
Liu RY et al (2014) Acute hyperglycemia together with hematoma of high-glucose blood exacerbates neurological injury in a rat model of intracerebral hemorrhage. Neurosci Bull 30(1):90–98
Zhang T et al (2019) Mitophagy reduces oxidative stress via Keap1 (Kelch-like epichlorohydrin-associated protein 1)/Nrf2 (nuclear factor-E2-related factor 2)/PHB2 (prohibitin 2) pathway after subarachnoid hemorrhage in rats. Stroke 50(4):978–988
Orekhov AN et al (2019) Mitochondrion as a selective target for treatment of atherosclerosis: role of mitochondrial DNA mutations and defective mitophagy in the pathogenesis of atherosclerosis and chronic inflammation. Curr Neuropharmacol
Li Q et al (2014) Rapamycin attenuates mitochondrial dysfunction via activation of mitophagy in experimental ischemic stroke. Biochem Biophys Res Commun 444(2):182–188
Zheng YQ et al (2009) RNA interference-mediated downregulation of Beclin1 attenuates cerebral ischemic injury in rats. Acta Pharmacol Sin 30(7):919–927
Zhang X et al (2014) Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: involvement of PARK2-dependent mitophagy. Autophagy 10(10):1801–1813
Zille M et al (2017) Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 48(4):1033–1043
Sun B et al (2018) Melatonin upregulates nuclear factor erythroid-2 related factor 2 (Nrf2) and mediates mitophagy to protect against early brain injury after subarachnoid hemorrhage. Med Sci Monit 24:6422–6430
Chude CI, Amaravadi RK (2017) Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int J Mol Sci 18(6):346
Castillo K et al (2013) Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis 4:e917
Gozuacik D, Kimchi A (2007) Autophagy and cell death. Curr Top Dev Biol 78:217–245
Acknowledgements
This work was funded by China Postdoctoral Science Foundation (2017M612010), National Natural Science Foundation of China (81701144, 81371433), National Key Research and Development Program of China (2017YFC1308500), and Key Program of Science and Technology Development of Zhejiang (2017C03021).
Author information
Authors and Affiliations
Contributions
WX, UO, and LG wrote the paper; ST drew the figures and tables; AS, CJL, and JZ revised the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors state that there was no conflict of interest in the preparation of this review.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, W., Ocak, U., Gao, L. et al. Selective autophagy as a therapeutic target for neurological diseases. Cell. Mol. Life Sci. 78, 1369–1392 (2021). https://doi.org/10.1007/s00018-020-03667-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-020-03667-9