
Abstract
In this chapter we review recent work towards high resolution imaging of unstained biological cells in the hydrated and living state, using synchrotron radiation (SR) and free electron laser (FEL) radiation. Specifically, we discuss the approaches of scanning small-angle X-ray scattering (scanning SAXS) and coherent diffractive X-ray imaging (CDI) of cells.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

Keywords
- scanning small-angle X-ray scattering (scanning SAXS)
- nano-diffraction
- coherent diffractive X-ray imaging (CDI)
- cytoskelleton
- microfluidics
1 X-ray Structure Analysis of Biological Cells: A Brief Overview
The desire to probe the three-dimensional (3D) structure of biological cells and tissues at high resolution and under hydrated conditions has motivated a continuous and long-lasting effort to develop suitable high resolution microscopy techniques. Fluorescence light microscopy provides an excellent tool to label specific biomolecules and organelles. As the last three decades have shown, an ever increasing number of imaging problems can be addressed by this specific labeling approach. However, not only the strength but also the limitation of this microscopy technique is linked to the selective imaging of a few components within the cells. Firstly, fluorescence microscopy of living cells can typically not be applied when transfection with fluorescent proteins is not possible or too invasive. Secondly, some questions in biology and biophysics cannot be answered from map** selected macromolecular components, but necessitate the visualization of the entire mass density distribution in the cell. In these cases high resolution images with quantitative mass or electron density contrast are needed, rather than the distribution of a selected label. Hard X-rays with multi-keV photon energies can contribute exactly this contrast mechanism related to the native electron density distributions in biological matter.
Apart from this aspect, other specific advantages of X-rays are: (i) a scalable resolution down to the X-ray wavelength of Å to nm, (ii) a kinematic nature of the scattering process enabling quantitative image analysis unaffected by multiple scattering, (iii) element specific contrast variation exploiting anomalous effects at absorption edges, (iv) compatibility with unsliced (three-dimensionally extended), unstained and hydrated specimens due to the large penetration depth. In this chapter we review recent studies of biological cells with hard X-rays. We focus on proof-of-concept experiments with micro- and nano-focused X-ray beams which have extended classical small-angle X-ray scattering (SAXS) to cellular imaging, combining real and reciprocal space. Classical SAXS is known as a structural technique for soft and biological matter, biomaterials and proteins which does not offer any real space information and can hardly be used on systems which are as heterogeneous as a biological cell. We also include coherent diffractive X-ray imaging (CDI) techniques as an X-ray imaging modality, which can complement scanning SAXS. We do no include, however, X-ray fluorescence microscopy of cells, which is by now quite well established, see [1].
The development of X-ray microscopy and imaging techniques has always been closely related to the availability of high brilliance radiation, provided by synchrotron radiation sources, and recently also by X-ray free electron lasers (FEL). Ultra-short and high brilliance FEL pulses may offer sharp still images of structure even of living cells, since the signal is recorded before structural changes occur by radiation damage. However, these very recent opportunities should not lead us to believe that imaging of cells with X-rays is an entirely new research topic. It has, in fact, already started with the pioneering work in the eighties both by the Göttingen group of G. Schmahl [2] and the Brookhaven group led by Kirz [3]. Biological microscopy with Fresnel zone plates in the so-called water window spectral range is by now a mature technique [4, 5]. In this chapter we restrict ourselves to the more recent developments of hard X-ray microscopy (i.e. photon energies above 5 keV).
In scanning SAXS, resolution in real space and reciprocal space is combined in a hybrid manner. This differs from other approaches which either reach high resolution in reciprocal space—based on diffraction averaging over a large ensemble such as in SAXS—or in real space based on inverting the diffraction pattern, e. g. by CDI. In fact, scanning SAXS with nano- or microbeams combines high resolution in reciprocal space (by analysis of the diffraction patterns and accounting for the available q-range) with resolution in real space on the order of the beam size. The method can hence probe local structures (in reciprocal space) in a range smaller than the beamsize down to the length scale given by the signal-to-noise cut-off. This cut-off depends on the degree of order in the sample and is typically intermediate between length scales of the organelle and the molecular constituents. At the same time, the resolution in real space is limited by the focal spot size. Scanning SAXS has been first demonstrated on biomaterial specimen such as wood and bone [6, 7], and then also for various tissue samples [8], with typical real space resolution values in the range of several microns. More recently, nano-focusing techniques (see Chap. 3) have made it possible to reach spot sizes well below 100 nm, based on reflective (mirrors, waveguides), refractive (compound refractive lenses) or diffractive optics (Fresnel zone plates, mutlilayer zone plates). Nano-focusing with X-rays is reviewed in [9], and also treated in advanced textbooks [10], while biological materials and cells imaged by diffraction and scattering have been previously reviewed in [11].
Direct imaging in real space, with a resolution below the X-ray spot size, is enabled by (far-field) CDI or (near-field) holographic techniques, see Chap. 2. For extended and non-compact objects such as cells, ptychographic CDI, or multi-plane holographic recordings are well suited to solve the phase problem, since the support constraint cannot be used. As in scanning SAXS, contrast is based on the native electron density distribution in hydrated biological cells. However, in contrast to scanning SAXS the specimen is directly imaged in real space. This is possible without any labeling, fixation, or staining. Within certain dose restrictions and for a short time span, CDI is also amenable to living cells [12]. The dose values to observe a time series on the same cell, however, are prohibitively high. For static images, a resolution below 100 nm is possible at synchrotron sources, while a range below 10 nm may be reachable by single ultra-short X-ray pulse using FEL radiation. In fact, the first ptychographic imaging of a cell already achieved 85 nm resolution on low contrast (unstained) bacterial cells at a fluence of \(10^7\) photons/µm\(^2\) [13, 14]. Extrapolating from these results and assuming a \(I \propto q^{-4}\) power law decay for the scattering intensity FEL pulses, delivering 10\(^{13}\,\)photons/µm\(^2\) in a time span below 50 fs, results in a resolution better than 3 nm. Actual experiments, however, are still about a factor of ten above this estimate, see for example [15], who have reported 2D reconstructions of projected electron density with 37 nm (half period) resolution for living bacterial cells.
As we will review here, recent work has now brought scanning SAXS and CDI of biological cells to the level where they can complement optical fluorescence and electron microscopy. In particular, they can ‘shed X-ray light’ on unlabeled cellular structures in cells by providing an electron density based contrast. Biomolecular assemblies can hence be investigated without slicing and staining, in fixed cells and—with restrictions—also in living cells [16,17,18]. This new “contrast mechanism” can possibly be useful for a very diverse range of problems. Here we name just a few examples, which may simply be closer to our perspective than others:
-
protein network architecture and the impact on cellular mechanics
-
protein filament bundling by cross-linking
-
force generation in cellular locomotion and muscle contraction
-
DNA compaction in the nucleus
-
amyloid aggregation.
After the following section, which addresses requirements of beam preparation and sample environments, we will first review scanning SAXS, followed by a section on direct imaging by ptychography and holography. We then address the topic of cellular imaging with FEL, and close with a section discussing multi-scale imaging, from the cellular to tissue level.
2 Methods: X-ray Optics and Sample Environment
2.1 Focusing Optics and Imaging Modalities
Imaging of cells with hard X-rays has been enabled only by the recent progress in X-ray optics and focusing, required to concentrate photons on a single cell or to specific regions within the cell. Since scattering of biological matter is typically weak, due to low-Z elements involved, beam preparation, cleaning of the beam path by apertures, efficient detection, and background subtraction are major issues. Finally, in situ optical microscopy is required to select scanning regions, perform alignments, and to monitor the cell with respect to radiation damage. The work reviewed here has been performed at synchrotron beamlines which combine these functionalities, notably ID13 of the European Synchrotron Radiation Facility (ESRF) in Grenoble, the cSAXS beamline of the Swiss Light Source, and the Göttingen Instrument for Nano-Imaging with X-rays (GINIX), installed at the P10 coherence beamline of the PETRA III storage ring at DESY in Hamburg. GINIX has been specifically designed for imaging of cells and tissues by holography and scanning SAXS [19]. Furthermore, it is fully compatible with tomography and also correlative optical microscopy [20].

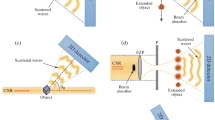
Adapted from [19]
Schematic of different imaging modalities for biological cells, for the example of the GINIX endstation of the P10 beamline (PETRAIII storage ring, DESY). a For scanning SAXS the beam is focused by Kikpatrick-Baez (KB) mirrors and cleaned by two successive soft-edge apertures (A1,A2) to cut the KB tails, and the diffraction is recorded by SAXS (D1) and WAXS (D2) detectors, each with respective beam stops (BS). b For ptychography, the KB-beam is made fully coherent by closing the entrance slits (SL), and the probe can be compactified by pinholes (P) if necessary for sampling. c For holographic imaging, the sample is moved to a defocus position, and after alignment with the pixel detector (D1), a high resolution detector (D4) is used to record the hologram. To increase the numerical aperture and hence the resolution, and to filter the wavefront, an X-ray waveguide (WG) is place into the focal plane of the KB. In this way, artifacts related to the typical wavefront distortions of a KB beam can be avoided.
As an example, Fig. 15.1 illustrates the different beam configurations and imaging modalities offered by the modular compound nano-focus optical system of GINIX. Similar modalities have also been realized at other beamlines. The optical system is composed of a high gain fixed curvature Kirkpatrick-Baez (KB) mirror and a probe filtering module, based on cleaning apertures and/or X-ray waveguides. Three different imaging modalities are sketched:
(a) Scanning SAXS, or more generally nano-diffraction in the small angle or wide-angle regime depending on detector position. For cells without mineralized or crystallized components, only SAXS signals are observed. Diffraction data are recorded for each scan point, forming a tensor product with two reciprocal space dimensions and two real space dimensions. As in conventional diffraction, a beamstop is required, sampling in real and reciprocal space is not very constrained, and coherence can be very low. The analysis is largely based on models and fitting of diffraction patterns in reciprocal space.
(b) Ptychography, i.e. far-field CDI with ptychographic phase retrieval. The slits in front of the KB are closed to achieve full coherence, and the anti-scatter apertures of (a) are replaced by pinholes to compactify the probe, i. e. to absorb the tails of the KB in focal space [21]. The sample is then scanned laterally behind the pinhole with partial overlap between exposures. Oversampling in the detector plane is required, and the beamstop must be sufficiently small or semi-transparent [22, 23] to recover low spatial frequencies.
(c) Holography, i.e. near-field phase contrast imaging in a diverging spherical wave, emitted from the exit of an X-ray waveguide. Due to the smaller confinement in the waveguide’s guiding channel, the divergence of the exit beam increases with respect to the KB beam, resulting in higher numerical aperture. The waveguide also results in wavefield and coherence filtering [24]. The holographic pattern is recorded by a high resolution detector, and is treated by phase retrieval in the optical near-field, as discussed in Chaps. 2 and 13. One advantage of (c) over (b) and (a) is that it is a full field technique and that images can be recorded without scanning. Both modalities (b, c) yield the projected electron density of the object.
2.2 X-ray Compatible Microfluidic Sample Environments for Cells
X-ray experiments on fixed hydrated and on living cells require a suitable sample environment, which closely mimics physiological conditions and does not further deteriorate the already low signal-to-noise ratios. For living cells, it is indispensable to provide nutrients and control metabolites by a continuous exchange media or buffer. Depending on the experiment, one may choose between cultivation of cells directly in the X-ray chamber or a suitable transfer strategy. Finally, a high throughput of suspended cells or the ability to scan a large number of adhering cells is important to obtain statistically relevant data sets. Another challenge to be faced when studying biological matter, and in particular living cells, is radiation damage. The dose is defined as
where \(I_0\) is the primary beam intensity in photons per time, T the exposure time, \(\rho \) the mass density of the sample, and \(\sigma \) the exposed area per scan point. Fast scanning, for example, helps to decrease the dose and protect the samples from deterioration.
To this end, the advent of microfluidic devices fabricated by soft lithography has been an enabling event for this research field. Even before X-ray experiments on cells had become a topic of interest, solution SAXS and structure analysis of suspended biomolecular assemblies had already been augmented by the possibility to observe in situ structural dynamics by making use of hydrodynamic focusing in microfluidic devices, as reviewed in [27, 28]. This approach paved the way to then adapt the fabrication processes and to develop X-ray compatible cell culture chambers [18, 25]. The most important requirements for flow chambers that are compatible with both X-ray studies and cell culture are: radiation stability, low absorption, low background scattering, control over the degree of cell adhesion to the materials, biological compatibility, and ease of fabrication. A method to custom-build flow chambers from UV curable adhesive as channel defining material, Kapton\(\tiny {\textregistered }\) film as radiation resistant window material and optional silicon-rich nitride (SiRN) windows as a substrate for cell growth is shown in Fig. 15.2a step-by-step and the resulting flow chambers are shown in photographs in Fig. 15.2b [25]. In these devices, the cells are constantly supplied with nutrients during the experiments and waste products of the cells are flushed away. Another advantage of the constant flow of liquid is that free radicals produced due to the radiation are flushed away, the sample is permanently cooled and air bubbles are reduced. The device design is very flexible since the fabrication is based on photolithography. Therefore, virtually any channel geometry can be realized and tested.

a Step-by-step sketch for fabricating X-ray compatible microfluidic devices for cell imaging. The insertion of the SiRN window is optional. b Photographs (back/front) of the flow chamber as described in (a). 1: PDMS (polydimethylsiloxane) support; 2: SiRN window; 3: flow channel; 4: inlet/outlet. c Flow chambers based on commercial microscopy slides with further insertion of a SiRN window. d Simple chambers with coverslip windows. a, b reproduced from [25] with permission from The Royal Society of Chemistry, c, d from [26]
In addition to the home-built flow chambers, one can use also commercially available microfluidic cell chambers (e.g. ibidi\(\tiny {\textregistered }\), Germany) as routinely used for optical microscopy. For X-ray microscopy, modified versions with adapted window materials (coated and uncoated SiRN) are preferred, see Fig. 15.2c. Finally, simple home-built chambers with windows made of cover slides may also do for fixed hydrated cells, if an absorption of X-ray photons by \(\le 200\) µm glass can be tolerated. In principle, a large set of window materials can be used for the X-ray measurements. The constraints differ for the imaging modality, since ‘low background’ in SAXS versus holography is associated with different material properties. For example, the residual phase shifts induced by traversing a few mms of Zeonor-\({\tiny \textregistered }\) were found to be small enough to still allow for phase contrast imaging of attached cells in the channel by holographic full-field imaging [12]. For scanning SAXS using a nanobeam, on the other hand, the excess polymer material resulted in an increased background level, which was reduced by inserting SiRN windows. As also compiled in a number of recent monographs [29,30,31,32,33], it is now fairly well known which window materials are compatible (and at which thickness) with a given imaging modality. Different window materials (glass, cyclic olefin copolymers [34], polypropylene) are now available for cellular growth.
An entirely different challenge is to create the sample environment for single pulse experiments by FEL. As a new sample has to be delivered to the ‘interaction zone’ for a subsequent ‘hit’, tailored microfluidic platforms are required. The strength of microfluidics is the high level of controllability. Flows are typically laminar, enabling exact control of important experimental parameters such as buffer/media conditions, temperature, induction period of reagents to the cells and so on. However, in contrast to synchrotron experiments, window-less flow chambers are necessary, since a single pulse would already destroy window materials. Free jets or microfluidic channels with holes offer of minimum background scatter and full compatibility with the X-ray beam propagating in ultra high vacuum.

a Microfluidic jet for sample delivery. Jets offer a windowless access to hydrated biomolecular samples, viruses, and bacteria with continuous high-rate replenishment of samples. In the diffraction experiment sketched, the jet (3) is aligned in the focal plane of a KB optic (1), behind a cleanup aperture (2), and the far-field diffraction is recorded by a 2D detector (5), with a miniaturized beamstop (4) blocking the primary beam directly behind the jet. b Finite element simulation of the flow field for a jet with diameter of 13 µm and a break-up length of a few mms. High flow verlocity can be used for orientational alignment of biomelecular assemblies, for example of a membrane suspension [35], but elongational and shear strain must be reduced for delivery of cells. c Photograph of the nozzle and the jet. d Optical stretcher. The sketch shows the integration of the laser system on the microfluidic chip, and the optical axis of the X-ray beam. The capillary is half-cut to give an inside view. The inset shows the central x-y-cross-section of the system. When a cell enters the trap, a highly anisotropic stress profile on the cell contour results in its trap** and stretching [36]. a–c adapted from [35], d from [37]
Figure 15.3a shows the example of a free microfluidic jet with laminar flow beneath a nozzle with \(13\,\upmu \)m exit, which has been used for diffraction on suspensions of biomolecules with micro-focus synchrotron radiation [35], but is also fully compatible with vacuum injection and therefore well suited for FEL experiments, see also [38]. The large speed of the jet, however, results in an elongational shear stress, which would certainly harm most eukaryotic cells, whereas it has been shown that bacteria can indeed be delivered by these jets, withstanding the hydrodynamic stresses [35]. In the meantime, jet technology has advanced tremendously, including electrostatic control, gas confinement and focusing layers [39]. Further, dedicated aerosole injectors have been designed for FEL sample delivery [40]. However, while some of these devices have been demonstrated to be suitable for viruses and smaller particles, the sample delivery for cells is still in its infancy. Living cells kept in a micro-liquid enclosure array, with each element used only once, as presented in [15], are one possible option. Jets with moderate hydrodynamic stress coupled out of microfluidic devices may offer more flexibility and control parameters. Finally, microfluidic channels with micro-sized holes drilled into their enclosure materials may be a third attractive route for further development.
Manipulation of cells in the beam and probing at controlled application of force is an entirely different challenge for the sample environment. In Fig. 15.3d we show the example of an optical stretcher, which was recently used to trap macrophages, and to rotate them in the beam for a tomographic scan [37]. In the stretcher, two opposing and divergent laser beams are used to trap and to stretch cells. The stretcher has been developed by Guck et al. [41] as a tool to studying the elastic properties of biological cells based on video microscopy of their deformed shape functions, since high deforming forces can be applied to biological objects such as cells. In this respect, stretchers offer an added functionality complementary to optical tweezers, which are commonly used to micro-manipulate micron and sub-micron-sized particles. In [37] the experimental capabilities of an optical stretcher as a potential sample delivery system for X-ray diffraction and imaging studies was explored. Even in a non-optimized configuration based on a commercially available optical stretcher system, X-ray holograms could be recorded from different views on a biological cell and the three-dimensional phase of the cell could be reconstructed. By means of high throughput screening, the optical stretcher could possibly become a useful tool, both for SR and FEL studies.
3 Scanning Small-Angle X-ray Scattering of Cells
In the past, we have focused our efforts to image cellular components mostly on, first, the cytoskeleton, due to particularly well ordered structures (see, e.g., Figs. 15.4a and 15.7a, b). Second, we have investigated the packing and (de-)compaction of DNA in the nucleus of eukaryotes (see Fig. 15.6a) and nucleoids of bacteria (Fig. 15.9d). The cytoskeleton determines the shape, motility, viscoelastic properties and generated forces of eukaryotic cells and is also a fascinating active soft matter system. It consist mainly of three distinct filament systems, along with associated binding proteins and molecular motors. Actin filaments play an essential role for directed cellular motion via polymerization and depolarization in lamellipodia and filopodia, form the stabilizing actin cortex and, together with myosin motors, stress fibers as the basic building blocks enabling the contractability of muscle cells. Microtubules contribute a system of ‘tracks’ within the cell, along which motor proteins transport cargo. They are also instrumental in cell division as they pull the chromosomes into the daughter cells. Intermediate filaments, such as vimentin or keratin, finally contribute greatly to passive mechanical properties of cells and protect the cell from destruction by heavy impact.
The DNA, which contains the genetic information of an organism, is densely packed in the nucleus or nucleoid and needs to be unpacked in a highly controlled manner for protein production. This apparent contradiction is solved by nature using a strictly hierarchical way of packing. To study the architecture and interactions of biomolecular assemblies, biophysicists have since long used SAXS. As the model systems get more and more complex, however, SAXS data interpretation is often impeded, in particular in the presence of large heterogeneity and the lack of molecule specific information available. Scanning SAXS, in particular when combined with visible light microsopy, addresses this challenge by providing additional real space constraints. Since scanning SAXS patterns average only locally over a small volume, a complete powder average is no longer obtained. For this reason, the anisotropy of the SAXS pattern can be measured, providing further clues. Of course, the indirect modeling of diffraction signals could be made obsolete altogether, if direct inversion of a coherent diffraction pattern was possible. However, this is to date not possible at the resolution which is typically achieved by SAXS. Apart from coherence and sampling issues which have to be met, inversion of data at higher momentum transfer and at low signal-to-noise level is much more difficult than just fitting a decay. Most importantly, model-based interpretation of SAXS data makes sense despite the fact that only one 2D projection is available. Contrarily, the information of the 2D projected electron density in real space, obtained for a single projection angle becomes useless at small scales without 3D tomography (which is often times technically not feasible).

Scanning SAXS with a nano-focused beam. a Inverted gray scale fluorescence micrograph of a SW13 cell with keratin K8/K18; the red arrow points to a particularly dense keratin bundle structure. b Corresponding X-ray dark field image overview image. c Composite image of individual SAXS patterns corresponding to the region in the red box in b. d Detailed X-ray dark field image of ROI (red box in b) recorded at smaller step size. e Single 2D diffraction pattern which clearly shows the orientation of the keratin bundles by the anisotropy of the signal (left) and integrated 1D intensity curve (right). Segments 1 and 5 align with the anisotropy and show distinct modulations of the signal. f X-ray darkfield scan of muscle induced human mesenchymal stem cell (hMSC), along with g the corresponding analysis of the anisotropy of the diffraction pattern revealing for example local orientations of networks of cytoskeletal components. h Single diffraction pattern of the region marked in (g). a–e adapted from [17], f–h from [26]
For this reason, we regard scanning SAXS as an indispensable tool in X-ray microscopy of biological cells, even if the first SAXS studies of individual cells (disregarding tissues and biomaterials) were only published in 2012 [17, 42]. One of the two papers used SAXS only as a complement to a ptychographic experiment to circumvent the stringent oversampling conditions, in search for structural clues on the condensation of DNA in the bacterial nucleoid [42]. Deinococcus radiodurans was chosen as a model system for the debated mechanisms of radiation damage repair [14, 42, 43]. Cells were prepared by rapid vitrification followed by freeze drying, and the SAXS signal was found to obey a power law decay with \(q^{-\nu }\) with \(\nu \simeq 3.2-3.7\), depending on the scan. The cross-over of the power law decay to the noise floor, which can serve as a resolution criterion, was observed at \(q=0.188\) Å\(^{-1}\). The first eukaryotic cells were studied at the same time by experiments dedicated to the development of scanning SAXS on cells. For this purpose, SW13 epithelial cells with a pronounced keratin K8/K18 network were chosen in order to highlight one of the three components of the cytoskeleton, see Fig. 15.4a–e. The cells were grown on SiRN windows, which are excellent substrates for cell growth and virtually transparent for X-rays, and subsequently chemically fixed, plunge-frozen and freeze-dried. This ensured a high electron density contrast between the cellular material and the surrounding air, which facilitated the recording in the starting phase. Cells were then imaged by scanning SAXS with a small beam (140 \(\times \) 110 nm\(^2\) and 200 \(\times \) 125 nm\(^2\)) and with small step sizes (50–2000 nm). For illustration, an X-ray dark field image, where in each position all scattering is integrated and plotted on a color scale, is shown in Fig. 15.4b, a detail in Fig. 15.4d. Example diffraction patterns are shown in Fig. 15.4c and at larger magnification in Fig. 15.4e. The signal was then integrated azimuthally to obtain 1D I(q) curves and, despite the small sample volume probed (beam size multiplied by sample thickness), distinct diffraction peaks were observed [17].
Pronounced streaks in diffraction patterns of Dictyostelium discoideum were explained with formation of fiber bundles in the acto-myosin contractile ring, based on comparing the ring like occurance of these features in SAXS scan to the typical contractile ring observed in fluorescence microscopy [16]. Modulations in the streaks were modeled based on a simple fiber scattering model [16, 32]. The strength and anisotropy of diffraction patterns were compared between naive human mesenchymal stem cells and differentiated stem cells, indicating that order of the cytoskeleton of stem cells increases during the differentiation process [26]. Modulations in the azimuthally integrated SAXS data could even be quantified by rendering the qualitative models into fitting procedures. Thus, the packing geometry, filament diameter and center-to-center distance and bundle diameter could be determined for keratin bundles in intact cells. The results correspond exactly to those derived from scanning electron microscopy, albeit without the need for slicing the cell [44].
Yet another highly ordered state of actin, namely hair cell stereocilia from the inner ear, were studied in [45]. These cell protrusions act as force sensors and enable hearing by transforming mechanical bending into a neuronal signal. They are filled with numerous parallel, densely packed actin filaments. Stereocilia were ‘stamped’ on coated SiRN windows, chemically fixed, stained for actin, and freeze-dried. Scanning SAXS experiments revealed an extremely high order within the stereocilia, but also considerable spatial heterogeneity in the translational and orientational structure.

Reprinted with permission from [18], Copyright (2014) by the American Physical Society
a Darkfield image of a chemically fixed, hydrated cell and b of a living cell. Note that in (b) several scan lines were skipped in order to account for the severe effects of radiation damage in this case. c 1D radial intensities plotted against the q values; note that the data for living cells (blue) are scaled by a factor of 10 for better visibility. d Power law exponents of chemically fixed (black) and living (blue) cells. The latter are consistently larger, indicating nanoscale changes. e Difference signal; data for fixed cells subtracted from data for living cells. Thus, values below 0 show structures emerging upon fixation, whereas values above 0 hint at structures that were destroyed by the fixation process.
After these early studies proved successful regarding interpretation and signal level, the next step was to investigate chemically fixed and living cells in hydrated state. Interestingly, distinct differences in the power-spectra of living and chemically fixed cells (see Fig. 15.5) were observed, pinpointing to both emerging and destroyed nanostructures upon chemical fixation [18]. This study illustrated very clearly the advantage of X-ray nano-imaging over other imaging methods. So far, X-ray imaging is the only way to directly compare fixed (or labeled, stained) and untreated whole cells since extensive sample preparation is not necessary for imaging. The results become particularly important now that fluorescence microscopy, where chemical fixation is a routine method, reaches well into the affected length scales (see top axis in Fig. 15.4e). In further studies, scanning SAXS was carried out on cryogenically preserved (i. e. vitrified) cells [16], which offers a larger range of dose before damage is observed. A direct comparison of the power law exponents for the decaying 1D SAXS curves shows that the results from cryo-preserved samples can be reproduced at room temperature, however, the primary beam needs to be attentuated and the exposure time decreased [46].
Following the first proof-of-concepts of scanning SAXS for biological cells, the optimization of imaging and analysis capabilities became important. To this end, several issues of optics, instrumentation and analysis were addressed: (i) choice of focus and influence of focal size on the SAXS data quality, (ii) rapid scanning using continuous movements and synchronized detector read-out, (iii) improvements of alignment procedures and tools based on in situ optical microscopy, (iv) optimized pixel detector read out and control software [47], (v) optimized beam preparation and suppression of tail scattering, for example, of the KB mirror system, (vi) specially fabricated semi-transparent beam stops [22], as well as (vii) the completion of a versatile toolbox for scanning SAXS which was made publically available [48, 49]. In this way, it now has become possible to compute structural observables in a fast and automated way, based on empirical data descriptors. Algorithms for semi-automatic quantification of the diffraction patterns include analysis of anisotropy parameters by automized fitting of ellipsoids [17], decomposition into principal components [26], the automized power law fitting [18, 44, 50, 51] and the computation of cumulants [50] to describe the azimuthally averaged structure factors for different regions-of-interest within the cells. In this way, empirical analysis has become possible also for extremely large data sets. At the same time, model based analysis of diffraction patterns, based on fiber models with free fitting parameters [16, 44], or sarcomere models for muscle cells [49] has been used. The effects of different sample preparation methods on the detected structure factor, including the freeze-dried, chemically fixed, frozen hydrated and living states was investigated more systematically. Radiation dose effects were also studied, and dose was precisely quantified by including ptychographic reconstruction of the probing beam [42]. In several studies, scanning SAXS and CDI was combined on the same cell, including combinations of SAXS with holography [51], and SAXS with ptychography [42, 44, 45].

Reprinted with permission from [46], Copyright (2016) by the American Chemical Society
a Schematic of the cell and nucleus division cycle of a eukaryotic cell. b Porod exponents of data taken from cells in different stages of the cell dividing cycle. An increase of the exponent towards 3.8 coincides with the decompaction of the DNA as the cell grows. The subsequent decrease is related to the compaction as the DNA is duplicated. Spatial representation of the DNA compaction state, c dark field image, d Porod exponent and e Porod constant. Interestingly, the Porod exponent is fairly homogeneous throughout nucleus and cytoplasm indicating a similar degree of heterogeneity in the structures. The Porod constant, by contrast, highlights dense reagins in the nucleus, presumably nucleoli.
It is not possible (yet) to record “movies” of living cells using scanning SAXS. Thus, to obtain temporally resolved information, an indirect approach by recording “snap shots” of cell has to be taken. An example is shown in Fig. 15.6, where the stage in the cell division cycle was determined by visible light bright field microscopy and the nuclei of these cells were recorded by scanning SAXS. Thus, it is possible to relate the SAXS signal and the exponents derived to the cell cycle time point (Fig. 15.6b) and derive information of the compaction and decompaction of the DNA.

From [50]
Scanning SAXS with a micro-focused beam. a, b Visible light fluorescence micrographs of a freeze-dried neonatal rat cardiac tissue cell with labeled actin cytoskeleton. The most significant filaments were identified by the filament sensor algorithm [52], see (a) (yellow lines), and a mean orientation angle was computed for all segmenting blocks with a blocksize equal to the stepsize of the SAXS scan, see (b) (red lines). c X-ray dark field of the SAXS scan. d PCA results on each diffraction pattern showing the fiber orientation as black lines. The degree of anisotropy was quantified by a unitless order parameter \(\omega _\mathrm{PA}\) as further detailed in [26, 50]. e Composite image showing the diffraction patterns with respect to their relative recording position. Black and gray arrows show the direction of the major and minor principal component axis. Diffraction patterns can be integrated azimuthally, see (e, inset), indicating the local structure factors, which can then be described mathematically by different fitting models (pink lines). Scale bar: 40 µm.
The best choice of real space and reciprocal space resolution in scanning SAXS deserves some special consideration, and a compromise has to be found for the specific application at hand. For example, a tight nano-focus leads to higher divergence and often also pronounced beam tails or streaks which can easily compromise the low-q signal in SAXS pattern. If this is to be prevented, the beam size must not be too small, typically in range of a few microns rather than in the range of a few hundred nanometers. Figures 15.7 and 15.8 show examples of micro-SAXS for cardiac tissue cell, where a clean recording of the sarcomere diffraction pattern at low-q required relaxation of real-space resolution to the micron range. Figure 15.7 shows that the corresponding real space images of neonatal rat cardiac tissue cells can still be recognized and correlated to the fluorescence micrographs, while at the same time the quality of the SAXS signal was improved, enabling automated decomposition of scattering patterns into principal components [26]. Using the micro-SAXS approach and hydrated adult cardiomyocytes, the myofibril diffraction signal reflecting inter-filament distances of thick filaments (myosin) and thin filaments (actin) could indeed be observed from selected regions within a single cell, see Fig. 15.8.

Adapted from [53]
Micro-SAXS results obtained for adult murine cardiomyocytes (CM). a Isolated adult CM from rat (optical brightfield image). b The diffraction signal recorded in each scan point reflects the acto-myosin structure of myofibrils. c The extracted d-spacing of the acto-myosin lattice, and d the filament orientation, determined by automated fitting. e Different physiological conditions of the heart and fixatives yield different interfilament periods. The distribution functions (histograms) of the structural parameters is reflected by the violin plots.
4 Coherent X-ray Imaging of Cells
4.1 Ptychography
In scanning SAXS, the real-space resolution is determined by the focal spot size. Contrarily, super-resolution below spot size can be achieved by CDI, where an oversampled far-field diffraction pattern is inverted by solving the phase problem. This was first achieved only under the restrictive assumption of a compact object with known support, fully coherent and plane wave illumination [54, 55]. The restriction to a ‘compact object’ was lifted by ptychographic CDI (pCDI), which uses a compact probe and partial overlap between illuminations of adjacent scan points to phase the diffraction pattern [56,57,58]. Exploiting the constraint of separability for phasing, both an unknown object o and an unknown probe p can be reconstructed [58], so that pCDI became applicable not only to extended samples, but also to non-idealized illumination functions (probes), including partial coherence [59]. A first application of ptychography to biological cells was shown in [14] for the gram-positive bacterium Deinococcus radiodurans [14]. Images of the projected mass density in freeze-dried preparations were reconstructed with a phase resolution below 0.01 rad up to a half-period resolution of 85 nm, at a relatively low fluence of \(6.6\times 10^7 \)photons\(/\upmu \)m\(^2\). Subsequent studies for the same bacteria, with substantially improved optics and phase retrieval extended this to 3D tomographic imaging [42, 43], see Fig. 15.9, and to cryogenically fixed cells [32]. By poviding mass density values, the results contributed to the long standing debate on DNA compactification in bacterial nucleoids [43]. In [60], malaria infected red blood cells were imaged by ptychographic tomography. In [61], quantitative phase and amplitude pCDI reconstructions were used to segment features and to localize mitochondrial constriction sites in mouse embryonic fibroblasts. Frozen-hydrated cells, which tolerate more dose than room temperature samples, and thus offer higher resolution, were imaged in 2D [62, 63], and in 3D by X-ray nano-tomography in [64]. Correlative cellular ptychography with functionalized Fe nanoparticles was shown in [65].

From [42]
Ptychographic reconstructions with simultaneous reconstruction of probe and object. a Phase image of a Siemens star test pattern with 50 nm lines and spaces, and b complex-valued field of the probe (focused beam) in the sample position. c Cuts along the optical axis of the propagated probe indicating significant astigmatism in the X-ray optics (Fresnel zone plate with upstream focusing). The results show that even with an astigmatic probe ptychography can yield faithful object reconstructions. d Tomographic reconstruction of Deinococcus radiodurans bacteria. Dense regions are attributed to DNA-rich bacterial nucleoids, obtained with the same setup at 6.2 keV photon energy (cSAXS beamline, Swiss Light Source).
4.2 Holography
The downsides of both original CDI and ptychography are: (i) stringent oversampling and coherence conditions, (ii) comparatively slow convergence of the reconstruction, (iii) rather long data acquisition for large field of view (no zooming out), and (iv) the high radiation dose. A major problem of applying CDI and pCDI to biological cells is in fact the low contrast in the hydrated state, and the correspondingly high radiation dose and associated radiation damage [66]. In fact, the typical radiation doses of CDI are in the range of \(10^7-10^9\) Gy, creating a need for cryogenic conditions to warrant structure preservation. The theoretical dose-resolution curve is characterized by an already very steep algebraic increase with an exponent \(3 \le \gamma \le 4\), as derived for the case of Fraunhofer far-field diffraction [66]. As an example, CDI was applied to hydrated bacterial cells (‘wet’ CDI), demonstrated at about 30 nm resolution (stated as half-period throughout this work), but at the ‘cost’ of \(10^8\) Gy [67].
These shortcomings could possibly be solved by holographic X-ray imaging, employing a highly coherent divergent cone for illumination and geometric adjustable magnification, and providing a much directed encoding of phase information. As shown in [12], X-ray waveguides can provide the clean wavefronts required for high resolution inline holography. While this approach has not yet reached the resolution of pCDI or CDI, it was proven experimentally to be very dose efficient. A dose advantage over CDI was also found by numerical simulations [68]. Furthermore, by variation of the object position, it becomes easily possible to zoom in an out and thus record multiple magnifications of a sample. Combined studies by holography and ptychography with scanning SAXS were published in [51] and [42, 44, 45], respectively. Holography, in particular, allows for low dose overviews recorded before identification of regions-of-interest for scanning SAXS. Figure 15.10 illustrates the state-of-art for holographic tomography of cells [69].

From [69]
Holo-Tomography reconstruction of a macrophage, labeled with barium sulfate and osmium tetroxide. a Virtual slice through a plane perpendicular to the tomographic axis. The cell with its internal contrast particles, the Petri dish and the resin used for embedding can be distinguished based on grey shades which are proportional to electron density. b Virtual slice coplanar to a projection direction. c 3D rendering of the dataset, showing barium particles (green) and the cell volume (half-transparent blue). Phase retrieval was based on the CTF approach with 4 distances. Scalebars denote 5 \(\upmu \)m.
By now the initially rather exotic area of CDI of cells has become more and more established. However, even ptychography, which is already more common than holography, has not yet spread into the biological community, for which many forms of microscopy are available. Contrarily, for tomography of biological materials and tissues it has found convincing use and for this purpose, the holographic approach has become more widespread than ptychography, see [70] for a state-of-the art application to human neuronal tissue. One very promising application of X-ray imaging on cells is the combination with other microscopy methods, as decribed in the next section.
5 Correlative Microscopy
Correlative X-ray and optical microscopy may help to overcome some of the persisting challenges in X-ray data analysis of scanning SAXS and will at the same time provide information not accessible by employing just one or the other method. In particular, correlative optical fluorescence microscopy can help to formulate models and constrain parameters, by providing additional information on specifically labeled biomolecules. In the absence of such information, previous studies of biological cells by scanning SAXS were mostly analyzed in terms in empirical, model-free data analysis, with only few exceptions [16, 44]. As shown in Sect. 15.3, already without a scattering model, a wealth of parameters can be extracted from the diffraction patterns in an automated manner, for example total diffraction intensity (darkfield), differential phase contrast, second moments of the scattering distribution, power-law exponents, or anisotropy parameters based on fitting of the 2D scattering patterns or principal component analysis (PCA) [17, 50]. By inspection of the diffraction patterns and the real-space maps, it seems plausible to attribute diffraction signals in some locations to the presence of filamentous proteins of the cytoskeleton or DNA in the nucleus. However, such conclusions need confirmation by optical fluorescence microscopy, at highest possible resolution. With this information at hand, the local diffraction patterns can be interpreted and analyzed, providing in the end much more information than either the optical image or the X-ray data alone.
In [20] a correlative microscopy approach for biological cells and tissues was proposed, which combines holographic X-ray imaging, X-ray scanning diffraction, and stimulated emission depletion (STED)-microscopy as a super-resolution optical fluorescence technique. All three imaging modalities were integrated into the same dedicated synchrotron nano-focus endstation GINIX at the P10 beamline of the PETRAIII storage ring (DESY, Hamburg). With this setup, both labeled and unlabeled biomolecular components in the cell can be imaged in a quasi-simultaneous scheme, exploiting the complementary contrast mechanisms of X-ray microscopy and optical fluorescence. This was demonstrated for heart tissue cells with a fluorescently labeled actin cytoskeleton. Micrographs of all three modalities were registered. The principal directions of the anisotropic diffraction patterns were found to coincide to a certain extent with the actin fiber directions. Further, actin filaments bundles were also recognizable in the phase map reconstructed from holographic recordings. We expect that the co-localization constraints provided by such correlative microscopy approaches will be instrumental for the formulation of advanced diffraction models, to fully exploit the data which is becoming available (Fig. 15.11).

From [20]
Correlative microscopy. Neonatal cardiac tissue cell with labeled actin, imaged in three different modalities. a STED micrograph. Scale bar: 5 µm. b X-ray phase reconstruction. c X-ray dark field map of the cell obtained by scanning SAXS.
6 From Cells to Tissues
For many biological functions it is important to integrate structural aspects on scales ranging from the single cell to the entire organ. Heart contractility as one of the most important physiological functions is a perfect example of how function relies on an intricate molecular and cellular architecture. The classical research field which addresses the way that cells form a functional tissue is histology, which combines sophisticated sectioning with optical or electron microscopy. While the cyto-architecture can thus be imaged in 2D, conventional histology lacks the capability to probe the tissue structure in full 3D. Furthermore, the high resolution molecular structure as revealed by electron microscopy can only be carried out in very small volumes, and structural variations within the tissues. Important and functionally relevant structural properties, such as for example the 3D vector field of myofibril orientation in heart, cannot be suitably assessed by conventional histology. Furthermore, different regions within the heart exhibit variations of the intrinsic sarcomere structure. For example, the acto-myosin lattice spacing near the ventrical wall may differ from the outer perimeter of the heart, as observed for mouse heart [49], see Fig. 15.12. Using the scanning SAXS approach it becomes possible to probe molecular orientation of heart tissue, combining the required real space resolution with molecular sensitivity by diffraction [49]. Extending this to a series of slices, or - as an alternative - to X-ray darkfield tomography [71], one could possibly probe the entire 3D assembly of myofibrils. In this way, the multi-scale challenge of map** molecular structures and orientation over length scales of an entire heart may become possible in future.

From [49]
Scanning SAXS of mouse cardiac muscle. a Optical micrograph of a histological section. b X-ray darkfield image, i.e. the integrated scattering intensity. c Multiple scattering parameters extracted in a fully automated manner from the diffraction patterns of the scan: (left) anisotropy of the scattering resulting from the (1, 1) reflection from the acto-myosin lattice, (center) the corresponding myofibril orientation, and (right) the mean position of the reflection along \(q_r\), as obtained from a Gaussian fit to the structure factor \(I(q_r)\) with a background model. Scale bar: 1mm.
Scanning small and wide-angle X-ray scattering (SAXS/WAXS) and X-ray fluorescence (XRF) with micro-focused synchrotron radiation have also been used in [72] to study histological sections from human brain tissue, notably of the midbrain and of substantia nigra. Both XRF and scanning SAXS/WAXS were shown to visualize tissue properties, which are inaccessible by conventional microscopy and histology. While scanning SAXS provided the local orientation and ordering of myelin structure, WAXS provide the distribution of cholesterol crystallites, and XRF maps of transition metals. All observables were intrinsically registered (aligned) since they were acquired in the same scan. Transition metals and more generally elemental distribution has become a relevant topic for neurodegeneration, for example the iron distribution and speciation in Parkinson’s disease (PD). In [72], variations in transition metal concentration between a PD and CTR patient were observed. The XRF analysis showed increased amounts of iron and decreased amounts of copper in the PD tissue compared to the control. PD tissue scans also exhibited increased amounts of crystallized cholesterol. However, as only tissues from one PD patient and one control were available, [72] can only serve as a proof-of-feasibility.
7 Outlook: FEL Studies of Cells
The advent of highly brilliant pulsed X-ray radiation from free electron lasers (FEL) has opened up a novel route to high resolution imaging by short femto-second (fs) pulses, before radiation damage takes place [73,74,75]. Ultra short pulses not only enable highest temporal resolution for example for pump-probe experiments, but also static single pulse imaging unaffected by any (Brownian) motion. If the structure is recorded by ultra-fast elastic scattering before changes occur due to multiple ionization, this holds promise to record sharp still images of extremely high resolution, unaffected by structure deterioration due to radiation damage. This so-called “diffract and destroy” principle was initially coined for single molecule CDI envisioned for FEL, but can also be applied to colloids, viruses or (small) entire cells. To this end, feasibility of imaging living cells by ultrafast CDI was discussed in [76], based on numerical simulations of the interaction of FEL pulses (10–100 fs) with biomolecular matter. It was concluded that subnanometer resolutions could be reached on micron-sized cells at fluences of \(10^{11}\)–\(10^{12} \)photons\(/\upmu \)m\(^2\). For mimivirus [77] and small bacteria of microbacterium lacticum [15], single pulse CDI has been indeed been demonstrated by now. However, resolution was much lower, i.e. 37 nm (half-period) for the bacterial cells.
Furthermore, single-shot CDI of large extended objects such as eukaryotes is in practice impeded by oversampling restrictions, the beam stop induced missing data problem [22], and the lack of a priori information (support). This is well illustrated by the FEL experiments on freeze dried cells presented below in Fig. 15.13. Notably, current pixel detector technology restricts the field of view to around \(d \simeq 1\upmu \)m for hard X-rays, which is prohibitive for eukaryotic cells. At the same time, FEL imaging of cells is limited to 2D. 3D imaging by serial shots with randomly sampled projections is possible for identical particles, but not for most cell types. A serial implementation of cellular imaging with high throughput of cells would nevertheless give a useful distribution of 2D views for a given cell type and state.
How can the maximum support cross section d resulting from support/oversampling constraints be increased? As we have \(d=\lambda z / 2 p\), where p is the detector pixel size, z the distance, and \(\lambda \) the wavelength, it seems reasonable to increase the wavelength for eukaryotic cells, which in addition also increases the scattering intensity. To this end, single pulse CDI experiments on freeze dried cells were also carried out in the water window spectral range, using FEL radiation of \(\lambda = 8\) mn at the FLASH facility (DESY, Hamburg). While the signal and hit rate were sufficient, see Fig.15.13, several restrictive conditions have impeded reconstruction: (1) Insufficient degree of coherence: The global degree of coherence of the third harmonic was determined to be only around 0.4 [78]. (2) Insufficient data at low q: The missing data due to overly sized beam stops and beam stop holders leads to unconstrained low frequencies. (3) Insufficient sampling: The large fields of view needed for the adherent cells were still not compatible with the oversampling conditions. From this attempt and other examples, one can learn that the strategy to collect and analyze data for eukaryotic cells at FEL has to be revised.

From D.-D. Mai et al., unpublished
Single pulse CDI experiments on cells, using soft X-ray FEL radiation at BL2/FLASH, DESY (\(\lambda = 8\) mn), at experimental parameters as reported in [78,79,80]. a Schematic of freeze dried cells attached to the multi-window SiRN array (576 100 \(\times \) 100 µm squared windows) with membrane thickness of 100 nm. The system is designed such that a single shot at a given micro-chamber will leave the other chambers with cells intact. b Optical microscopy of the D. radiodurans (top) and SK8K18 (human epithelial) cells (bottom). c Setup with the focused beam, multi-window sample holder, and the thin foils used to suppress the 3rd harmonic at \(\lambda _{3\mathrm{rd}}= 2.66\) nm by a 200 nm free-standing Pd-filter, installed directly in front of the CCD detector. To avoid radiation damage on the CCD detector, a beam stop blocks the central pixels. d Single pulse diffraction patterns of D. radiodurans bacteria and human epithelial cell SK8K18. The magnification shows that the oversampling criterion is fulfilled at this wavelength. The power spectral density shows a dynamic range of more than three orders of magnitude, and hence a sufficient signal. However, the existence of a beam stop and the lack of compact support impedes object reconstruction.
The solution to this challenge could be a hybrid approach based on combining two separate paradigms of structural analysis: imaging and diffraction. Low and medium resolution projected electron density should be assessed in real space, based on near-field (holographic) phase retrieval, while high resolution (molecular) Fourier components should be assessed by model based analysis of diffraction data, relaxing the restrictive conditions for inversions. Importantly, by holographic (near-field) imaging—with the sample in the defocus position of the nano-focused FEL—even extended objects (without support constraints) can be reconstructed from single shot data. To this end, iterative algorithms using mild constraints can be readily employed in the near-field regime, such as negativity of the phase, unit amplitude (pure phase object) or sparsity, as well as combinations thereof. This hybrid approach could be implemented for example at the MID instrument at XFEL, using a compound refractive lens system focusing to 50 nm. At the same time, and in addition to the high resolution CCD or CMOS detectors needed for this imaging modality, a wide angle pixel detectors could be used to record the far field scattering intensity outside of the central diverging radiation cone, i. e. covering Fourier components corresponding to scales below 50 nm. Figure 15.14 shows a sketch of an FEL holography experiment. The sample is placed in controlled defocus position \(z_1\) behind the focal plane of the CRL. The direct beam traverses a pixel detector with a hole, and reaches the high resolution detector at large distance where the in-line hologram is recorded.

a Schematic of divergent FEL beam, calculated in natural units (Rayleigh length \(z_R\), waist \(w_0\)) for 8 keV photon energy, and beam focusing by 50 compound refractive lenses (CRL) made of Beryllium (\(R=50\) µm, \(f=94.6\) mm, \(w_0=45.9\) nm); parameters are adapted to MID instrument of XFEL. Data can be recorded in two ways: (i) single shot far-field diffraction patterns recorded by a pixel detector (AGIPD) at 10 m distance, and (ii) holographic recordings by a high resolution sCMOS detector with the sample in a defocus position. b Simulation of the beam intensity at \(z_1=5\) mm behind the CRL focus, overlaid with the cell phantom. Scale bar 2.5 µm. c Simulated hologram at 10 m distance. Scale bar 1 mm
References
de Jonge, M.D., Vogt, S.: Hard X-ray fluorescence tomography—an emerging tool for structural visualization. Curr. Opin. Struct. Biol. 20(5), 606–614 (2010). https://doi.org/10.1016/j.sbi.2010.09.002. https://doi.org/10.1016/j.sbi.2010.09.002
Schmahl, G., Rudolph, D., Niemann, B., Christ, O.: X-ray microscopy of biological specimens with a zone plate microscope. Ann. NY Acad. Sci. 342(1), 368–386 (1980)
Jacobsen, C., Kenney, J., Kirz, J., Rosser, R., Cinotti, F., Rarback, H., Pine, J.: Quantitative imaging and microanalysis with a scanning soft X-ray microscope. Phys. Med. Biol 32(4), 431 (1987)
Larabell, C.A., Nugent, K.A.: Imaging cellular architecture with X-rays. Curr. Opin. Struct. Biol. 20(5), 623–631 (2010)
Schneider, G., Guttmann, P., Heim, S., Rehbein, S., Mueller, F., Nagashima, K., Heymann, J.B., Müller, W.G., McNally, J.G.: Three-dimensional cellular ultrastructure resolved by X-ray microscopy. Nat. Meth. 7(12), 985 (2010)
Lichtenegger, H., Müller, M., Paris, O., Riekel, C., Fratzl, P.: Imaging of the helical arrangement of cellulose fibrils in wood by synchrotron X-ray microdiffraction. J. Appl. Cryst. 32(6), 1127–1133 (1999)
Rinnerthaler, S., Roschger, P., Jakob, H., Nader, A., Klaushofer, K., Fratzl, P.: Scanning small angle X-ray scattering analysis of human bone sections. Calcified Tissue Int. 64(5), 422–429 (1999)
Bunk, O., Bech, M., Jensen, T., Feidenhans, R., Binderup, T., Menzel, A., Pfeiffer, F.: Multimodal X-ray scatter imaging. New. J. Phys. 11(12), 123,016 (2009)
Attwood, D., Sakdinawat, A.: X-rays and Extreme Ultraviolet Radiation: Principles and Applications. Cambridge University Press (2017)
Stangl, J., Mocuta, C., Chamard, V., Carbone, D.: Nanobeam X-ray Scattering: Probing Matter at the Nanoscale. Wiley (2013)
Hémonnot, C.Y.J., Köster, S.: Imaging of biological materials and cells by X-ray scattering and diffraction. ACS Nano 11(9), 8542–8559 (2017). https://doi.org/10.1021/acsnano.7b03447. https://doi.org/10.1021/acsnano.7b03447
Bartels, M., Krenkel, M., Haber, J., Wilke, R., Salditt, T.: X-ray holographic imaging of hydrated biological cells in solution. Phys. Rev. Lett. 114(4), 048,103 (2015)
Giewekemeyer, K., Neubauer, H., Kalbfleisch, S., Krüger, S.P., Salditt, T.: Holographic and diffractive X-ray imaging using waveguides as quasi-point sources. New J. Phys. 12(3), 035,008 (2010)
Giewekemeyer, K., Thibault, P., Kalbfleisch, S., Beerlink, A., Kewish, C., Dierolf, M., Pfeiffer, F., Salditt, T.: Quantitative biological imaging by ptychographic X-ray diffraction microscopy. Proc. Natl. Acad. Sci. U.S.A. 107, 529 (2010)
Kimura, T., Joti, Y., Shibuya, A., Song, C., Kim, S., Tono, K., Yabashi, M., Tamakoshi, M., Moriya, T., Oshima, T., et al.: Imaging live cell in micro-liquid enclosure by X-ray laser diffraction. Nat. Commun. 5, 3052 (2014)
Priebe, M., Bernhardt, M., Blum, C., Tarantola, M., Bodenschatz, E., Salditt, T.: Scanning X-ray nanodiffraction on dictyostelium discoideum. Biophys. J. 107(11), 2662–2673 (2014)
Weinhausen, B., Nolting, J.F., Olendrowitz, C., Langfahl-Klabes, J., Reynolds, M., Salditt, T., Köster, S.: X-ray nano-diffraction on cytoskeletal networks. New J. Phys. 14(8), 085,013 (2012)
Weinhausen, B., Saldanha, O., Wilke, R.N., Dammann, C., Priebe, M., Burghammer, M., Sprung, M., Köster, S.: Scanning X-ray nanodiffraction on living eukaryotic cells in microfluidic environments. Phys. Rev. Lett. 112(8), 088,102 (2014). https://doi.org/10.1103/PhysRevLett.112.088102
Salditt, T., Osterhoff, M., Krenkel, M., Wilke, R.N., Priebe, M., Bartels, M., Kalbfleisch, S., Sprung, M.: Compound focusing mirror and X-ray waveguide optics for coherent imaging and nano-diffraction. J. Synchrotron Radiat. 22(4), 867–878 (2015)
Bernhardt, M., Nicolas, J.D., Osterhoff, M., Mittelstädt, H., Reuss, M., Harke, B., Wittmeier, A., Sprung, M., Köster, S., Salditt, T.: Correlative microscopy approach for biology using X-ray holography, X-ray scanning diffraction and sted microscopy. Nat. Commun. 3641 (2018)
Giewekemeyer, K., Wilke, R.N., Osterhoff, M., Bartels, M., Kalbfleisch, S., Salditt, T.: Versatility of a hard X-ray kirkpatrick-baez focus characterized by ptychography. J. Synchr. Radiat. 20(3), 490–497 (2013)
Wilke, R., Vassholz, M., Salditt, T.: Semi-transparent central stop in high-resolution X-ray ptychography using kirkpatrick-baez focusing. Acta Cryst. A 69(5), 490–497 (2013)
Wilke, R., Wallentin, J., Osterhoff, M., Pennicard, D., Zozulya, A., Sprung, M., Salditt, T.: High-flux ptychographic imaging using the new 55 \(\upmu \)m-pixel detectorlambda’based on the medipix3 readout chip. Acta Cryst. A 70(6), 552–562 (2014)
Osterhoff, M., Salditt, T.: Coherence filtering of x-ray waveguides: analytical and numerical approach. New J. Phys. 13(10), 103,026 (2011)
Weinhausen, B., Köster, S.: Microfluidic devices for x-ray studies on hydrated cells. Lab Chip (2013). https://doi.org/10.1039/c2lc41014a
Bernhardt, M., Priebe, M., Osterhoff, M., Wollnik, C., Diaz, A., Salditt, T., Rehfeldt, F.: X-ray micro- and nanodiffraction imaging on human mesenchymal stem cells and differentiated cells. Biophys. J. 110(3), 680–690 (2016)
Ghazal, A., Lafleur, J.P., Mortensen, K., Kutter, J.P., Arleth, L., Jensen, G.V.: Recent advances in x-ray compatible microfluidics for applications in soft materials and life sciences. Lab Chip 16(22), 4263–4295 (2016). https://doi.org/10.1039/c6lc00888g. https://doi.org/10.1039/c6lc00888g
Köster, S., Pfohl, T.: X-ray studies of biological matter in microfluidic environments. Mod. Phys. Lett B 26(26), 1230,018 (2012)
Bernhardt, M.: X-ray micro- and nano-diffraction imaging on human mesenchymal stem cells and differentiated cells. Ph.D. thesis, Georg-August-Universität Göttingen (2017)
Hemonnot, C.: Investigating cellular nanoscale with x-rays. Ph.D. thesis, Georg-August-Universität Göttingen (2016)
Nicolas, J.D.: Mulitscale x-ray analyis of biological cells and tissues by scanning diffraction and coherent imaging. Ph.D. thesis, Georg-August-Universität Göttingen (2018)
Priebe, M.: Scanning x-ray nanodiffraction on dictyostelium discoideum. Ph.D. thesis, Georg-August-Universität Göttingen (2015)
Weinhausen, B.: Scanning x-ray nano-diffraction on eukaryotic cells: From freeze-dried to living cells. Ph.D. thesis, Georg-August-Universität Göttingen (2013)
Denz, M., Brehm, G., Hémonnot, C.Y.J., Spears, H., Wittmeier, A., Cassini, C., Saldanha, O., Perego, E., Diaz, A., Burghammer, M., Köster, S.: Cyclic olefin copolymer as an x-ray compatible material for microfluidic devices. Lab Chip 18(1), 171–178 (2018). https://doi.org/10.1039/c7lc00824d. https://doi.org/10.1039/c7lc00824d
Priebe, M., Kalbfleisch, S., Tolkiehn, M., Köster, S., Abel, B., Davies, R., Salditt, T.: Orientation of biomolecular assemblies in a microfluidic jet. New J. Phys. 12(4), 043,056 (2010)
Lincoln, B., Wottawah, F., Schinkinger, S., Ebert, S., Guck, J.: High-throughput rheological measurements with an optical stretcher. Meth. Cell Biol. 83, 397–423 (2007)
Nicolas, J.D., Hagemann, J., Sprung, M., Salditt, T.: The optical stretcher as a tool for single-particle x-ray imaging and diffraction. J. Synchrotron Radiat. 25(4) (2018)
DePonte, D., Weierstall, U., Schmidt, K., Warner, J., Starodub, D., Spence, J., Doak, R.: Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D: Appl. Phys. 41(19), 195,505 (2008)
Chavas, L., Gumprecht, L., Chapman, H.: Possibilities for serial femtosecond crystallography sample delivery at future light sources. Struct. Dynam. 2(4), 041,709 (2015)
Kirian, R., Awel, S., Eckerskorn, N., Fleckenstein, H., Wiedorn, M., Adriano, L., Bajt, S., Barthelmess, M., Bean, R., Beyerlein, K., et al.: Simple convergent-nozzle aerosol injector for single-particle diffractive imaging with x-ray free-electron lasers. Struct. Dynam. 2(4), 041,717 (2015)
Guck, J., Ananthakrishnan, R., Mahmood, H., Moon, T.J., Cunningham, C.C., Käs, J.: The optical stretcher: a novel laser tool to micromanipulate cells. Biophys. J. 81(2), 767–784 (2001)
Wilke, R., Priebe, M., Bartels, M., Giewekemeyer, K., Diaz, A., Karvinen, P., Salditt, T.: Hard x-ray imaging of bacterial cells: nano-diffraction and ptychographic reconstruction. Opt. Express 20(17), 19232–19254 (2012)
Wilke, R.: Coherent x-ray diffractive imaging on the single-cell-level of microbial samples: ptychography, tomography, nano-diffraction and waveguide-imaging. Ph.D. thesis, Georg-August-Universität Göttingen (2014)
Hemonnot, C.Y., Reinhardt, J., Saldanha, O., Patommel, J., Graceffa, R., Weinhausen, B., Burghammer, M., Schroer, C.G., Köster, S.: X-rays reveal the internal structure of keratin bundles in whole cells. ACS Nano 10(3), 3553–3561 (2016)
Piazza, V., Weinhausen, B., Diaz, A., Dammann, C., Maurer, C., Reynolds, M., Burghammer, M., Köster, S.: Revealing the structure of stereociliary actin by x-ray nanoimaging. ACS Nano 8(12), 12228–12237 (2014)
Hémonnot, C.Y.J., Ranke, C., Saldanha, O., Graceffa, R., Hagemann, J., Köster, S.: Following DNA compaction during the cell cycle by x-ray nanodiffraction. ACS Nano 10(12), 10661–10670 (2016). https://doi.org/10.1021/acsnano.6b05034. https://doi.org/10.1021/acsnano.6b05034
Osterhoff, M.: dada–a web-based 2d detector analysis tool. J. Phys. Conf. Ser. 849, 012059 (2017) (IOP Publishing)
Nicolas, J.D., Bernhardt, M., Markus, A., Alves, F., Burghammer, M., Salditt, T.: Scanning x-ray diffraction on cardiac tissue: automatized data analysis and processing. J. Synchrotron Rad. 24(6), 1163–1172 (2017). https://doi.org/10.1107/S1600577517011936. https://doi.org/10.1107/S1600577517011936
Bernhardt, M., Nicolas, J.D., Eckermann, M., Eltzner, B., Rehfeldt, F., Salditt, T.: Anisotropic x-ray scattering and orientation fields in cardiac tissue cells. New J. Phys. 19(1), 013,012 (2017)
Nicolas, J.D., Bernhardt, M., Krenkel, M., Richter, C., Luther, S., Salditt, T.: Combined scanning x-ray diffraction and holographic imaging of cardiomyocytes. J. Appl. Cryst. 50(2), 612–620 (2017). http://journals.iucr.org/j/issues/2017/02/00/rg5124/rg5124.pdf
Eltzner, B., Wollnik, C., Gottschlich, C., Huckemann, S., Rehfeldt, F.: The filament sensor for near real-time detection of cytoskeletal fiber structures. PLoS ONE 10(5), e0126,346 (2015)
Nicolas, J.D., Bernhardt, M., Schlick, S.F., Tiburcy, M., Zimmermann, W.H., Khan, A., Markus, A., Alves, F., Toischer, K., Salditt, T.: X-ray diffraction imaging of cardiac cells and tissue. Prog. Biophys. Mol. Biol. (2018)
Chapman, H.N., Barty, A., Marchesini, S., Noy, A., Hau-Riege, S.P., Cui, C., Howells, M.R., Rosen, R., He, H., Spence, J.C., et al.: High-resolution ab initio three-dimensional x-ray diffraction microscopy. JOSA A 23(5), 1179–1200 (2006)
Miao, J., Charalambous, P., Kirz, J., Sayre, D.: Extending the methodology of x-ray crystallography to allow imaging of micrometre-sized non-crystalline specimens. Nature 400(6742), 342 (1999)
Rodenburg, J., Hurst, A., Cullis, A., Dobson, B., Pfeiffer, F., Bunk, O., David, C., Jefimovs, K., Johnson, I.: Hard-x-ray lensless imaging of extended objects. Phys. Rev. Lett. 98(3), 034,801 (2007)
Rodenburg, J.M.: Ptychography and related diffractive imaging methods. Adv. Imag. Electr. Phys. 150, 87–184 (2008)
Thibault, P., Dierolf, M., Menzel, A., Bunk, O., David, C., Pfeiffer, F.: High-resolution scanning x-ray diffraction microscopy. Science 321(5887), 379–382 (2008)
Thibault, P., Menzel, A.: Reconstructing state mixtures from diffraction measurements. Nature 494(7435), 68 (2013)
Jones, M.W., Van Riessen, G.A., Abbey, B., Putkunz, C.T., Junker, M.D., Balaur, E., Vine, D.J., McNulty, I., Chen, B., Arhatari, B.D., et al.: Whole-cell phase contrast imaging at the nanoscale using fresnel coherent diffractive imaging tomography. Sci. Rep. 3, 2288 (2013)
Jones, M.W., Elgass, K., Junker, M.D., Luu, M.B., Ryan, M.T., Peele, A.G., Van Riessen, G.A.: Map** biological composition through quantitative phase and absorption x-ray ptychography. Sci. Rep. 4, 6796 (2014)
Deng, J., Vine, D.J., Chen, S., Nashed, Y.S., **, Q., Phillips, N.W., Peterka, T., Ross, R., Vogt, S., Jacobsen, C.J.: Simultaneous cryo x-ray ptychographic and fluorescence microscopy of green algae. Proc. Natl. Acad. Sci. U.S.A. p. 201413003 (2015)
Lima, E., Diaz, A., Guizar-Sicairos, M., Gorelick, S., Pernot, P., Schleier, T., Menzel, A.: Cryo-scanning x-ray diffraction microscopy of frozen-hydrated yeast. J. Microsc. 249(1), 1–7 (2013)
Diaz, A., Malkova, B., Holler, M., Guizar-Sicairos, M., Lima, E., Panneels, V., Pigino, G., Bittermann, A.G., Wettstein, L., Tomizaki, T., et al.: Three-dimensional mass density map** of cellular ultrastructure by ptychographic x-ray nanotomography. J. Struct. Biol. 192(3), 461–469 (2015)
Gallagher-Jones, M., Dias, C.S.B., Pryor, A., Bouchmella, K., Zhao, L., Lo, Y.H., Cardoso, M.B., Shapiro, D., Rodriguez, J., Miao, J.: Correlative cellular ptychography with functionalized nanoparticles at the fe l-edge. Sci. Rep. 7(1), 4757 (2017)
Howells, M.R., Beetz, T., Chapman, H.N., Cui, C., Holton, J., Jacobsen, C., Kirz, J., Lima, E., Marchesini, S., Miao, H., et al.: An assessment of the resolution limitation due to radiation-damage in x-ray diffraction microscopy. J. Electron Spectrosc. 170(1–3), 4–12 (2009)
Nam, D., Park, J., Gallagher-Jones, M., Kim, S., Kim, S., Kohmura, Y., Naitow, H., Kunishima, N., Yoshida, T., Ishikawa, T., et al.: Imaging fully hydrated whole cells by coherent x-ray diffraction microscopy. Phys. Rev. Lett. 110(9), 098,103 (2013)
Hagemann, J., Salditt, T.: The fluence-resolution relationship in holographic and coherent diffractive imaging. J. Appl. Cryst. 50(2), 531–538 (2017)
Krenkel, M., Toepperwien, M., Alves, F., Salditt, T.: Three-dimensional single-cell imaging with x-ray waveguides in the holographic regime. Acta Cryst. A 73(4), 282–292 (2017)
Töpperwien, M., van der Meer, F., Stadelmann, C., Salditt, T.: Three-dimensional virtual histology of human cerebellum by x-ray phase-contrast tomography. Proc. Natl. Acad. Sci. U.S.A. p. 201801678 (2018)
Liebi, M., Georgiadis, M., Menzel, A., Schneider, P., Kohlbrecher, J., Bunk, O., Guizar-Sicairos, M.: Nanostructure surveys of macroscopic specimens by small-angle scattering tensor tomography. Nature 527(7578), 349–352 (2015). https://doi.org/10.1038/nature16056. https://doi.org/10.1038/nature16056
Carboni, E., Nicolas, J.D., Töpperwien, M., Stadelmann-Nessler, C., Lingor, P., Salditt, T.: Imaging of neuronal tissues by x-ray diffraction and x-ray fluorescence microscopy: evaluation of contrast and biomarkers for neurodegenerative diseases. Biomed. Opt. Express 8(10), 4331 (2017). https://doi.org/10.1364/boe.8.004331. https://doi.org/10.1364/boe.8.004331
Chapman, H.N., Fromme, P., Barty, A., White, T.A., Kirian, R.A., Aquila, A., Hunter, M.S., Schulz, J., DePonte, D.P., Weierstall, U., et al.: Femtosecond x-ray protein nanocrystallography. Nature 470(7332), 73 (2011)
Gaffney, K., Chapman, H.: Imaging atomic structure and dynamics with ultrafast x-ray scattering. Science 316(5830), 1444–1448 (2007)
Neutze, R., Wouts, R., van der Spoel, D., Weckert, E., Hajdu, J.: Potential for biomolecular imaging with femtosecond x-ray pulses. Nature 406(6797), 752 (2000)
Bergh, M., Huldt, G., Timneanu, N., Maia, F.R., Hajdu, J.: Feasibility of imaging living cells at subnanometer resolutions by ultrafast x-ray diffraction. Q. Rev. Biophys. 41(3–4), 181–204 (2008)
Seibert, M.M., Ekeberg, T., Maia, F.R., Svenda, M., Andreasson, J., Jönsson, O., Odić, D., Iwan, B., Rocker, A., Westphal, D., et al.: Single mimivirus particles intercepted and imaged with an x-ray laser. Nature 470(7332), 78 (2011)
Mai, D., Hallmann, J., Reusch, T., Osterhoff, M., Düsterer, S., Treusch, R., Singer, A., Beckers, M., Gorniak, T., Senkbeil, T., et al.: Single pulse coherence measurements in the water window at the free-electron laser flash. Opt. Express 21(11), 13005–13017 (2013)
Dronyak, R., Gulden, J., Yefanov, O., Singer, A., Gorniak, T., Senkbeil, T., Meijer, J.M., Al-Shemmary, A., Hallmann, J., Mai, D., et al.: Dynamics of colloidal crystals studied by pump-probe experiments at FLASH. Phys. Rev. B 86(6), 064,303 (2012)
Singer, A., Sorgenfrei, F., Mancuso, A., Gerasimova, N., Yefanov, O., Gulden, J., Gorniak, T., Senkbeil, T., Sakdinawat, A., Liu, Y., et al.: Spatial and temporal coherence properties of single free-electron laser pulses. Opt. Express 20(16), 17480–17495 (2012)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2020 The Author(s)
About this chapter

Cite this chapter
Salditt, T., Köster, S. (2020). Scanning Small-Angle X-ray Scattering and Coherent X-ray Imaging of Cells. In: Salditt, T., Egner, A., Luke, D.R. (eds) Nanoscale Photonic Imaging. Topics in Applied Physics, vol 134. Springer, Cham. https://doi.org/10.1007/978-3-030-34413-9_15
Download citation
DOI: https://doi.org/10.1007/978-3-030-34413-9_15
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-030-34412-2
Online ISBN: 978-3-030-34413-9
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)