
Abstract
Background
While polyploids are common in plants, the evolutionary history and natural dynamics of most polyploid groups are still unclear. Owing to plentiful earlier systematic studies, Ludwigia sect. Isnardia (comprising 22 wetland taxa) is an ideal allopolyploid complex to investigate polyploid evolution and natural dynamics within and among taxa. With a considerable sampling, we concentrated on revisiting earlier phylogenies of Isnardia, reevaluating the earlier estimated age of the most recent common ancestor (TMRCA), exploring the correlation between infraspecific genetic diversity and ploidy levels, and inspecting interspecific gene flows among taxa.
Results
Phylogenetic trees and network concurred with earlier phylogenies and hypothesized genomes by incorporating 192 atpB-rbcL and ITS sequences representing 91% of Isnardia taxa. Moreover, we detected three multi-origin taxa. Our findings on L. repens and L. sphaerocarpa were consistent with earlier studies; L. arcuata was reported as a multi-origin taxon here, and an additional evolutionary scenario of L. sphaerocarpa was uncovered, both for the first time. Furthermore, estimated Isnardia TMRCA ages based on our data (5.9 or 8.9 million years ago) are in accordance with earlier estimates, although younger than fossil dates (Middle Miocene). Surprisingly, infraspecific genetic variations of Isnardia taxa did not increase with ploidy levels as anticipated from many other polyploid groups. In addition, the exuberant, low, and asymmetrical gene flows among Isnardia taxa indicated that the reproductive barriers may be weakened owing to allopolyploidization, which has rarely been reported.
Conclusions
The present research gives new perceptions of the reticulate evolution and dynamic nature of Isnardia and points to gaps in current knowledge about allopolyploid evolution.
Similar content being viewed by others


Background
Polyploidization has long been recognized as an essential force in plant evolution (Raven and Thompson 1964; Leitch and Bennett 1997; Soltis et al. 2014); however, the complex nature of polyploids has restrained research efforts on these plants (Soltis et al. 2009; Dufresne et al. 2014; Baduel et al. 2018). Many questions about polyploid evolution are not yet thoroughly resolved. For example, how did polyploidization occur? How many times has polyploidization occurred in the history of a specific group? Do polyploids interact with closely related taxa; if yes, how? Is a polyploid taxon diverse at the molecular level; if yes, how? An increasing number of studies are attempting to dissect the origins of polyploids and the dynamic nature among and within polyploid taxa by applying the rapidly develo** molecular and computational techniques (e.g. Van de Peer et al. 2021; Karbstein et al. 2022; Zhuang et al. 2022). Ludwigia L. section Isnardia (L.) W.L. Wagner & Hoch, a polyploid complex with a moderate number of species and sufficient knowledge from plenty of earlier systematic studies, is an ideal plant group to explore polyploid phylogeny as well as the dynamic nature of polyploids.
Isnardia currently contains 22 taxa, including five diploids, 11 tetraploids, four hexaploids, one octoploid, and one taxon with an unknown ploidy level (Table 1) (Raven 1963; Raven and Tai 1979; Peng 1988, 1989; Peng et al. 2005; Wagner et al. 2007; Arya et al. 2020; Liu et al. 2020). Isnardia are wetland weeds characterized by their cree** or erect stem, 4-merous and haplostemonous flowers, terete or globose capsules, and pluriseriate and free seeds (Wagner et al. 2007). Members of Isnardia were previously classified into three sects. Isnardia, Michelia Ramamoorthy, and Microcarpium Munz (Munz 1944; Raven 1963; Ramamoorthy 1979; Wagner et al. 2007). Early systematic studies have suggested that these three sects. are closely related and, together, compose the Microcarpium complex (Eyde 1977, 1978, 1981; Peng and Tobe 1987; Peng et al. 1988, 2005; Tobe et al. 1988; Peng 1989; Wagner et al. 2007). Based on morphological and anatomic studies, this complex was proposed to be evolutionarily closed to sect. Ludwigia L. (Eyde 1977, 1978, 1981). Recent molecular phylogenies revealed that this complex is a monophyletic group with strong (Liu et al. 2020) or weak (Liu et al. 2017) support, and sect. Ludwigia is sister to the complex (Liu et al. 2017, 2020). However, none of the three sects. in the complex exhibit monophyly (Hung et al. 2009; Liu et al. 2017, 2020), therefore the three sects. in the complex were combined into the sect. Isnardia (Liu et al. 2020). Examining the combination with different data sets will provide valuable information on this taxonomic treatment.
Extant Isnardia plants are mainly distributed in North America with two exceptions––L. ovalis Miq. is endemic to East Asia and L. venugopalanii S. Arya, V. Suresh, P. Biju & V.S.A. Kumar occurs in India––and one widely spread taxon, L. palustris (L.) Elliott, which occurrs in North, Central, and South America, Eurasia, and Africa (Raven 1963; Peng et al. 2005; Wagner et al. 2007; Arya et al. 2020). Geographical distribution and phylogeographic analyses indicated that Isnardia plants originated in North America and a few taxa spread to other continents later (Raven 1963; Eyde 1981; Tobe et al. 1988; Liu et al. 2020). The earliest known fossil record of Isnardia indicated that this clade had extended to Europe in Middle Miocene (Friis 1985; Tobe et al. 1988) (ca. 11.63–15.97 million years ago (MYA) referring to the International Commission on Stratigraphy (ICS), https://stratigraphy.org/). That is, Isnardia likely originated before Middle Miocene. However, the results of Hung et al. (2009)’s coalescence analyses suggested a younger the most recent common ancestor (TMRCA) of Isnardia, which was 5.99 ± 0.02 MYA based on chloroplast atpB-rbcL data and 6.59 ± 0.02 MYA based on nuclear ITS data. Additional molecular data, more thorough sampling, and further analyses are warranted to give a better perception of the divergence time of Isnardia.
Origins of Isnardia taxa have been intensively investigated using morphological, anatomic, cytological, and molecular data (Raven 1963; Eyde 1977, 1978, 1981; Raven and Tai 1979; Peng and Tobe 1987; Tobe et al. 1988; Peng 1988, 1989; Peng et al. 2005; Hung et al. 2009; Liu et al. 2017, 2020). Eight ancestral genomes––including genomes A, B (= H), C (= I), D (= G), F1, F2, M1, and M2––have been proposed and assigned to each Isnardia taxon (Table 1) based on cytological and molecular works (Peng 1988, 1989; Peng et al. 2005; Hung et al. 2009; Liu et al. 2020). The reticulate origins of the 16 allopolyploid Isnardia––consisting of 11 tetraploids, four hexaploids, and one octoploid (Table 1)––were also inferred (Liu et al. 2020). Multiple origins are common in polyploid speciation (Soltis and Soltis 1993; Leitch and Bennett 1997; Peng and Chiang 2000; Doyle et al. 2004). To date, however, only two Isnardia––tetraploid L. sphaerocarpa Elliott and hexaploid L. repens J.R. Forst.––have been considered to have multiple origins (Peng 1988; Liu et al. 2020), because of the limitation of small sample sizes for each taxon in the earlier studies. Comprehensively decoding the polyploid Isnardia taxa's origins requires a larger sample size for each taxon.
In addition, higher genetic diversities have been detected in the taxa with higher ploidy levels in several plant groups (e.g. Mallet 2007; García‐Verdugo et al. 2009; Bogačiovienė et al. 2019; Zhang et al. 2019). At the same time, earlier studies have observed plenty of natural hybrids among Isnardia taxa in the field and reported the high capability of interspecific hybridization among Isnardia taxa in breeding experiments at research greenhouses (Raven and Tai 1979; Peng 1988, 1989; Peng et al. 2005). However, these attributes of Isnardia taxa have not been well explored. Hung et al. (2009) measured infraspecific genetic diversities of six Isnardia taxa. Both Liu et al. (2017) and Liu et al. (2020) sampled 20 Isnardia taxa, but they could not approximate interspecific gene flow nor infraspecific genetic diversities due to small sample sizes for each taxon. Moreover, a larger sample size for each taxon will provide a better insight into these characteristics of Isnardia.
In the present study, we intended to meet the following four aims by comprehensively sampling Isnardia. First, we reexamined the reticulate evolution hypotheses implied by earlier studies (Peng 1988, 1989; Peng et al. 2005; Hung et al. 2009; Liu et al. 2020) and investigated multiple origins of polyploid taxa. Second, we reevaluated the estimated ages of Isnardia TMRCA (Hung et al. 2009). Third, we tested the proposition that, as with many other allopolyploid plant groups (Mallet 2007; García‐Verdugo et al. 2009; Bogačiovienė et al. 2019; Zhang et al. 2019), genetic diversities of Isnardia taxa increase with their ploidy levels. Fourth, consistent with earlier field observations and breeding experiments (Raven and Tai 1979; Peng 1988, 1989; Peng et al. 2005), we investigated the interspecific gene flows among Isnardia taxa with different ploidy levels.
Methods
Sampling
We aimed to sample all Isnardia taxa. For each Isnardia taxon, we planned to sample three to ten individuals. Living plants were collected during collecting trips in Alabama, Florida, Massachusetts, Missouri, North Carolina, Oklahoma, South Carolina, Tennessee, Texas, and Virginia in the USA. Leaf tissue for genomic DNA extraction was dried in silica gel immediately after being collected. Vouchers were deposited at the Herbarium of Endemic Species Research Institute (TAIE) (Thiers 2016), Taiwan, for further studies. Samples of the Cuban endemic species, L. stricta (C. Wright ex Griseb.) C. Wright, were obtained from the herbarium vouchers provided by the Herbarium of Missouri Botanical Garden (MO) (Thiers 2016), Saint Louis, Missouri, USA. Unfortunately, samples for the recently described Indian endemic species, L. venugopalanii (Arya et al. 2020), were unavailable. Additional Isnardia taxa from Hung et al. (2009) were also incorporated into this study. In addition, published DNA sequences of some outgroups from other sections in Ludwigia and other genera in Onagraceae were downloaded from the GenBank (Sayers et al. 2019) for the analyses.
Genomic DNA extraction, PCR, cloning, sequencing, and assembly
The genomic DNA of our samples was extracted by optimizing the cetyltrimethylammonium bromide (CTAB) method (Murray and Thompson 1980; Doyle and Doyle 1987) for Isnardia samples. A mixture of 5–10 mg ground leaf tissue, 10 mL 65 °C 3X CTAB isolation buffer, and 40 μL 0.4% ß-mercaptoethanol was incubated at 65 °C for 30 min. After the incubation, 10 mL chloroform-isoamyl alcohol (24:1) was added to the mixture. The solution was gently, thoroughly mixed, and then centrifuged at 6000×g for 10 min at about 20 °C. The supernatant was retained, and the process described in the previous sentence was repeated twice. The final supernatant was mixed well with 10 mL isopropanol, incubated at − 20 °C for 30 min to 24 h, warmed up, and centrifuged at 15,000×g for 10 min at 4 °C. The precipitated DNA was air-dried, dissolved with 500 μL TE buffer, and incubated at 37 °C for 30 min with RNase A. A 500 μL isopropanol was applied to precipitate DNA again. The precipitated DNA was then resuspended with 70% ethanol, centrifuged at 15,000×g for 2–3 min at 4 °C, air-dried, and dissolved in 200 μL TE buffer for the following polymerase chain reactions (PCR).
Both nuclear ITS (ITS1 + 5.8S + ITS2) and chloroplast atpB-rbcL regions were amplified with the universal primers (White et al. 1990; Chiang et al. 1998; Hung et al. 2009) for all Isnardia samples. A 100 μL mixture including 0.5 μL 10U/μL Taq Polymerase (Promega, Madison, Wisconsin, USA), 10 μL 10X PCR buffer, 10 μL 8 mM dNTP, 10 μL 2 pM forward primer, 10 μL 2 pM reverse primer, 10 μL 10 mM MgCl2, 10 μL 2 ng/ μL genomic DNA, and distilled water was applied in each PCR amplification. The genomic DNA in the mixture was initially denatured at 92 °C for 5 min, followed by 31 cycles of 92 °C for 45 s, 53 °C for 75 s, and 72 °C for 90 s, and finally elongated at 72 °C for 10 min. The PCR products were then purified with 1% agarose gel and the Gel/PCR DNA Isolation System (Viogene, Taipei, Taiwan).
In the case where multiple sizes of PCR products were amplified for one individual, TA cloning was applied using the pGEM-T Easy Cloning Vector (Promega, Madison, Wisconsin, USA) to determine variations within the individual.
Subsequently, PCR products and colonies were sequenced on an ABI 3730XL DNA Analyzer (Applied Biosystems, Waltham, Massachusetts, USA) commercially with the universal primers (White et al. 1990; Chiang et al. 1998; Hung et al. 2009). DNA reads were assembled using the De Novo Assemble tool implemented in Geneious Prime 2022.2.1 (Biomatters, Ltd., Auckland, New Zealand). Assembled DNA sequences were deposited at GenBank for further studies.
Sequence alignment, genetic variation, phylogenetic analysis, and network
DNA sequences for ITS and atpB-rbcL regions were aligned with Clustal Omega 1.2.3 (Sievers and Higgins 2014). To understand the infraspecific genetic variations of Isnardia taxa, the nucleotide diversities (Tajima’s π; π) (Tajima 1983) were quantified using MEGA 11 (Tamura et al. 2021) with the Kimura 2-parameter model (Kimura 1980), and 500 replicates were applied to calculate the standard errors. One-way analysis of variance (ANOVA) and student’s t-tests were conducted with SPSS 28.0 (IBM Corp 2021) to test our third hypothesis as well as to assess how sample size affects the genetic diversities of Isnardia.
To reconstruct phylogenetic trees and meet our first goal, we applied Maximum likelihood (ML) and Bayesian inference (BI) algorithms. The nucleotide substitution model for each studied region was obtained using jModelTest 2 (Darriba et al. 2012). The ML trees were generated using RAxML 8.2.11 (Stamatakis 2014) with the best-fitting model for each region, and the branch supports were evaluated using the bootstrap** values (bs) (Felsenstein 1985) with 1000 replicates. Bayesian inference (BI) analyses of phylogeny were conducted using MrBayes 3.2.7 (Huelsenbeck and Ronquist 2001; Ronquist et al. 2012) on the CIPRES Science Gateway 3.3 (Miller et al. 2010) with two independent Markov Chain Monte Carlo (MCMC) runs, 5 × 106 generation Markov chains in each run, and the best-fitting models. Trees were saved every 1000 generations. A 50% majority-rule consensus BI tree and the posterior probabilities (pp) on the branches were yielded by incorporating the last 75% of the saved trees. FigTree 1.4.4 (Rambaut 2018) was applied to depict the ML and BI trees.
To infer the evolutionary relationships within Isnardia, which includes many allopolyploid taxa, a phylogenetic network was conducted. The biparentally inherited nuclear regions are required in the network analyses to take in both maternal and paternal heritages of allopolyploid organisms. Therefore, a subset of the ITS data was applied here. Based on the Isnardia genomes hypothesized by Liu et al. (2020), in the subset, ideally, one sequence would be selected to represent a diploid taxon, two sequences from two different well-supported clades would represent a tetraploid taxon, three sequences from three different well-supported clades would represent a hexaploid, and so on. Here, we randomly selected one sequence to represent a taxon in each well-supported clade from the ITS tree generated above in the present study and made these sequences the subset. The subset was then aligned and analyzed using Clustal Omega 1.2.3 (Sievers and Higgins 2014) and RAxML 8.2.11 (Stamatakis 2014) as described above. The resulting subset ML tree was then converted to a multi-label tree. Subsequently, a phylogenetic network was computed with the multi-label tree algorithm using the Exact Method (Huber et al. 2006) implemented in Dendroscope 3.8.3 (Huson and Scornavacca 2012).
Divergence time estimation
To achieve our second aim, the age of Isnardia TMRCA was estimated using Beast 2.6.7 (Bouckaert et al. 2019) based on the subset ITS data and outgroup sequences from other Ludwigia sections and other genera in Onagraceae (see Additional file 1). Sequences were aligned with Clustal Omega 1.2.3 (Sievers and Higgins 2014), and the best-fitting nucleotide substitution model was determined using jModelTest 2 (Darriba et al. 2012). Applying the birth–death skyline model (BDSKY; Stadler et al. 2013), we ran a Markov Chain length of 4 × 107 generations with the estimated best-fitting nucleotide substitution model, substitution rates of ITS in genus Lopezia under Onagraceae (5.15 × 10–9 substitutions/site/year; O’Kane 1993), and secondary calibration points at the crown node of Onagraceae (71 (88.4–54.3) MYA from Gonçalves et al. (2020) and 46.9 (74.1–2.7) MYA from Zhang et al. (2021)). These two age estimations were both in harmony with those from the fossil records (Grímsson et al. 2011; Lee et al. 2013; Farooqui et al. 2019). Trees were sampled every 1,000 generations. Output log files were checked with Tracer 1.7.1 (Rambaut et al. Full size image

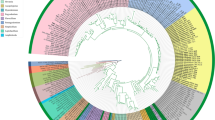
Maximum likelihood tree of Ludwigia sect. Isnardia inferred from the chloroplast atpB-rbcL region. The arrow indicates the crown node of Isnardia. Sample numbers (Additional file 1), ploidy levels (Table 1), and genome types (Table 1) are shown right after the taxa. Numbers at nodes show the bootstrap** values (bs) / posterior probabilities (pp) only when the bs at nodes are greater than 50 or pp at nodes are greater than 0.70. The scale bar denotes the branch length
Moreover, our phylogenetic analyses showed that both tetraploid L. sphaerocarpa (AABB) and hexaploid L. repens (AABBCC) have evolved multiple times, and tetraploid L. arcuata Walter (BBCC) might have multiple origins or a higher infraspecific genetic variation. For L. sphaerocarpa, one evolutionary scenario showed that the paternal and/or maternal donors of L. sphaerocarpa samples 1–3 and 5 were phylogenetically close to L. lanceolata Elliott (AABB) and L. suffruticosa Walter (AABB), two members of the AABB tetraploids (Fig. 1; Table 1; Additional file 1). This evolutionary scenario might fit earlier hypotheses on the homoploid hybridization of L. sphaerocarpa (Peng 1988; Liu et al. 2020) if our atpB-rbcL tree (Fig. 2) had higher resolution and gave more information on the maternal donors of these four L. sphaerocarpa samples. We detected another evolutionary scenario that revealed that the maternal donor of L. sphaerocarpa sample 4 probably carried genome A and was likely closely related to L. lanceolata and L. glandulosa Walter subsp. glandulosa (AABB), which both had an AABB genome type (Fig. 2; Table 1). The paternal donor contributing genome B to this L. sphaerocarpa sample likely also gave rise to L. curtissii Chapm. (BBCCDDF1F1) and L. simpsonii Chapm. (BBCCDD) (Fig. 1; Table 1). The latter evolutionary scenario of L. sphaerocarpa has not been revealed by earlier studies.
For the origins of L. repens, our atpB-rbcL tree (Fig. 2) suggests that the maternal donors were probably tetraploids carrying genome BBCC and closely related to L. arcuata. This result was congruent with earlier studies (Hung et al. 2009; Liu et al. 2020). Moreover, our ITS tree (Fig. 1) indicated that at least two ancestor lineages––likely the maternal donors with genome BBCC––had given birth to L. repens; one gave rise to L. repens sample 4 while another contributed to L. repens samples 1–3 and 5–9. The former maternal donor also played a part in the origination of L. brevipes (Long) Eame (AABBCC). However, no paternal donor of L. repens with genome A was represented in our study.
Our ITS tree shows that L. arcuata samples are clustered in two well-supported groups (Fig. 1). One group consists of L. arcuata samples 2–4, L. brevipes samples 1–6, and L. repens sample 4, while another group includes L. arcuata sample 1 and L. repens samples 1–3 and 5–9. This indicates that L. arcuata probably has multiple origins and/or a relatively high infraspecific nucleotide diversity. In fact, our analyses show that the infraspecific π of L. arcuata was higher than that of most Isnardia (Table 1).
Twenty-nine Isnardia samples were included in the subset ITS data and network analyses. Sample information of the subset is provided in Additional file 1. The phylogenetic network (Fig. 3) concurs with our ITS tree (Fig. 1) completely and indicates the hybrid origins of eight Isnardia taxa. Tetraploids L. lanceolata, L. pilosa Walter, and L. suffruticosa share the same evolutionary histories, and all of them are allopolyploids deriving from two genomes (Fig. 3). One evolutionary signal arose from the lineage sister to the clade/genome A. Another signal is likely from genome B because this signal arose from the lineage sister to L. ravenii C.I Peng, which has B in its genome type. Our ITS and atpB-rbcL trees (Figs. 1, 2) indicate that tetraploid L. sphaerocarpa had evolved at least twice (see previous paragraphs). The first evolutionary scenario, homoploid hybridization (Peng 1988; Liu et al. 2020), could not be shown in the network due to the limitation of our sampling. The second evolutionary scenario is revealed in our network (Fig. 3); that L. sphaerocarpa obtained genome A from the lineage sister to the A clade/genome and obtained its genome B from L. curtissii, which also has genome B. Moreover, two evolutionary signals were detected for the origins of tetraploid L. arcuata and hexaploid L. repens, and these two evolutionary signals were from sister lineages, which both lineages might have genome B (dashed red lines in Fig. 3). This may indicate that both L. arcuata and L. repens have multiple origins. Furthermore, our network implies that hexaploid L. simpsonii stemmed from at least two genomes––D genome from the lineage close to diploid L. microcarpa and B genome from the lineage close to L. curtissii. In addition, two evolutionary signals were mixed to give rise to L. ovalis. However, it was difficult to distinguish M1 and M2 signals in our network. Alignment, the ML tree, and network files are given in Additional file 4.

Evolutionary network of Ludwigia sect. Isnardia inferred from the subset of the ITS region. The arrow indicates the crown node of Isnardia. The A, M1M2, F1F2, and D clades are marked as in Fig. 1. Blue lines show the evolutionary signal from the A genome, red lines from the B genome, dashed red lines likely from the B genome, green lines from the D genome, brown lines from the M1 and M2 genomes
Furthermore, the results of our genetic variation analyses of Isnardia suggest that the atpB-rbcL region generally has lower infraspecific genetic variations (π = 0.00000–0.01896) compared to the ITS region (π = 0.00000–0.02097). The infraspecific π of each Isnardia taxon are shown in Table 1. Notably, ITS sequences are infraspecific identical in L. microcarpa Michx. and L. spathulata Torr. & A. Gray, and atpB-rbcL sequences are infraspecific identical in L. linifolia Poir. and L. microcarpa. One-way ANOVA analyses showed that the genetic diversities of Isnardia taxa had no significant differences among sample sizes (p-values > 0.05; Additional file 5). In other words, our sampling was appropriate for assessing the genetic diversities in Isnardia. Furthermore, our study detected no relationship between ploidy levels and genetic diversities (Fig. 4). For instance, the π value of the ITS region for diploid L. palustris (AA) was 0.00189 ± 0.00115, but some of the tetraploids with the AABB genome type had lower π values and others had higher π values (Table 1). One-way ANOVA analyses indicated no significant differences in π values of Isnardia taxa among ploidy levels (p-values > 0.05; Fig. 4; Additional file 5). Student’s t-tests also showed no significant differences in π values between diploids and all polyploids (p-values = 0.81 and 0.08 for ITS and atpB-rbcL regions, respectively). That is, our results falsified hypothesis three.

Boxplots showing infraspecific nucleotide diversities (Tajima’s π) of different ploidy levels in Ludwigia sect. Isnardia based on (a) atpB-rbcL and (b) ITS data. Letters above each boxplot indicate pairwise statistical differences between ploidy levels. The complete details for the statistical analysis are shown in Additional file 5. The infraspecific nucleotide diversity of each taxon is available in Table 1
Divergence time estimation
All (29) Isnardia samples in the subset and nine outgroups were applied to estimate the age of Isnardia TMRCA. Sample information is provided in Additional file 1. The topologies of the resulted chronograms (Fig. 5) are congruent with our ITS tree (Fig. 1). Adopting the secondary calibration points at the crown node of Onagraceae suggested by Zhang et al. (2021), the crown node of genus Ludwigia was approximately 17.2 (20.3–13.2) MYA, the crown node of the North Temperate Haplostemonous (NTH) Ludwigia (= sects. Isnardia + Ludwigia) was 7.6 (10.5–5.3) MYA, and TMRCA of Isnardia was 5.9 (7.7–4.3) MYA (Fig. 5a). When we took Gonçalves et al. (2020)’s results as calibration points instead, the crown node of genus Ludwigia was estimated to be 25.9 (30.5–20.2) MYA, the crown node of the NTH Ludwigia was 11.3 (15.7–7.8) MYA, and TMRCA of Isnardia was 8.9 (11.5–6.5) MYA (Fig. 5b). Alignment and 50% majority-rule consensus tree files are shown in Additional file 6.

Chronograms of Ludwigia sect. Isnardia based on the subset of ITS region with divergence time estimates adopting the substitution rates of ITS (O’Kane 1993) and secondary calibration point at the crown node of Onagraceae following (a) Zhang et al. (2021) or (b) Gonçalves et al. (2020). The arrows indicate the crown nodes of Isnardia. Sample numbers (Additional file 1), ploidy levels (Table 1), and genome types (Table 1) are shown right after the taxa. Stars at nodes indicate the posterior probabilities (pp) only if the pp at the nodes is greater than 0.90. When the pp at the nodes is greater than 0.95, the double star notations are denoted. Numbers at nodes show the median estimated divergence times (MYA) with 95% credible intervals in the brackets. The smears around the nodes display the uncertainty of the estimated divergence times
IMa analyses
The migration rates per gene copy per generation (M, interspecific gene flow) for every two Isnardia taxa based on ITS and atpB-rbcL regions revealed rampant but low interspecific gene flow in Isnardia, from 9.42 × 10–10 to 3.13 × 10–9, and 2.36 × 10–10 to 6.58 × 10–10, respectively (Fig. 6, Additional file 7). Moreover, our ANOVA and t-tests showed no significant differences in M among ploidy levels based on atpB-rbcL data but found some significant differences in ITS data (Fig. 6, Additional file 8). The M between two ploidy levels––except the M from hexaploids to diploids (group 6 × > 2 ×) and from hexaploids to octoploids (group 6 × > 8 ×) based on ITS data––were compatible with the M between two diploids (group 2 × > 2 ×) (see Fig. 6, Additional file 8). Moreover, Fig. 6b shows that the means of Ms from hexaploids to other ploids (including groups 6 × > 2 × , 6 × > 4 × , 6 × > 6 × , and 6 × > 8 ×) were lower than the means of Ms of other groups. Our data supported hypothesis four and indicated that Ms from hexaploids to other taxa were relatively lower.

Boxplots showing the estimated migration rates (M, migration rates per gene copy per generation) between ploidy levels in Ludwigia sect. Isnardia based on (a) atpB-rbcL and (b) ITS data. Groups on the x axial are coded, showing the direction of the migration rates, e.g., 2 × > 4 × indicates the migration rates from diploids to tetraploids. Letters above each boxplot indicate pairwise statistical differences (p-values < 0.05) between 2 × > 2 × and other groups. The asterisks indicate pairwise statistical differences (p-values < 0.05) between the two groups. The complete details for the statistical analysis are shown in Additional file 8. The estimated migration rates among taxa are available in Additional file 7
Furthermore, unsymmetrical Ms were detected in most two-taxon pairs (Additional file 7), and ITS data gave more information on the natural dynamic in Isnardia (Fig. 6b, Additional file 8a). Both the M of group 2 × > polyploid were significantly greater than the M of group polyploid > 2 × . In addition, our ITS data indicated that the M of group 4 × > 4 × was considerably higher than the Ms of groups 4 × > 2 × , 4 × > 6 × , and 4 × > 8 × , and hexploids had the same trend but no statistical significance. Diploids, however, showed an opposite trend without statistical significance. The M of group 2 × > 2 × was lower than the Ms of groups 2 × > 4 × and 2 × > 6 × , but higher than the M of group 2 × > 8 × .
