
Abstract
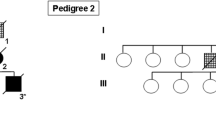
We describe an autopsied patient with familial parkinsonism and unclassified four repeat-tau (4R-tau) aggregation. She presented with bradykinesia, truncal dystonia, and mild amnesia at the age of 61 and then exhibited body weight loss (15 kg over 8 months), sleep disturbances, and progressive respiratory failure with CO2 narcosis. She died of respiratory failure at the age of 62, 14 months after disease onset. Her brother also showed parkinsonism at the age of 58 and suddenly died 6 months later. Postmortem examination revealed 4R-tau aggregation, which was characterized by neuronal globose-type tangles or pretangles, bush-like or miscellaneous astrocytic inclusions, and coiled bodies. The temporal tip, the striatum, the substantia nigra, the tegmentum of the midbrain, the medullary reticular formation, and the spinal cord were severely involved with tau aggregation. Argyrophilic grains and ballooned neurons were also found in the medial temporal structures, however, extensions of the 4R-aggregations in the case were clearly broader than those of the argyrophilic grains. Western blot analysis of sarkosyl-insoluble fractions from brain lysates revealed prominent bands of tau at both 33 kDa and 37 kDa. Genetic examinations did not reveal any known pathogenic mutations in MAPT, DCTN-1, PSEN-1, or familial or young-onset parkinsonism-related genes. The clinical manifestations, pathologic findings, and biochemical properties of aggregated tau in our patient cannot be explained by argyrophilic grain disease or other known 4R-tauopathies alone. Our results further extend the clinical and neuropathologic spectra of 4R-tauopathy.
Similar content being viewed by others

Introduction
Neuronal and glial aggregation of tau protein is known to cause neurodegenerative diseases. Tau protein is classified into three-repeat tau (3R-tau) and four-repeat tau (4R-tau), which are determined by alternative mRNA splicing of exon 10 [1, 10]. Aggregations of 4R-tau are pathologic hallmarks of corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), argyrophilic grain dementia (AGD), globular glial tauopathy (GGT), aging-related tau astrogliopathy (ARTAG), and microtubule-associated protein tau (MAPT) gene mutation-associated familial frontotemporal dementia (FTD) and parkinsonism, whereas neuronal inclusions of 3R-tau are characteristic of Pick’s disease (PiD) [17, Neuropathologic findings The brain weighed 1260 g before a fixation. The temporal tip and the hippocampus were mildly atrophied. The tegmentum of the brainstem, the medullary reticular formation, and the fasciculus anterolateralis of the spinal cord showed marked atrophy. The substantia nigra showed severe discoloration, whereas the locus coeruleus was retained. The cerebral neocortices, basal ganglia, and cerebellum were not atrophied (Fig. 2a–g). Microscopically, neuronal loss and astrogliosis were prominent in the temporal tip, amygdala, ambient gyrus, and substantia nigra (Fig. 2h–i). Occasional ballooned neurons were observed in those regions and in the anterior horn of the spinal cord (Fig. 2j, k). Immunohistochemistry revealed AT8- and RD4-immunopositive neuronal and glial inclusions to be abundant in the temporal tip, amygdala, CA1, subiculum, ambient gyrus, substantia nigra, tegmentum of the midbrain, reticular formation of the medulla oblongata, hypoglossal nerve nucleus, and anterior horn of the spinal cord (Fig. 3a–e). The neuronal inclusions were globose-type tangles or pretangles (Fig. 3f–g), and a subset of neuronal inclusions in the spinal anterior horn showed fine, fibrous configurations (Fig. 3h). The astrocytes demonstrated miscellaneous aggregations of hyperphosphorylated tau; bush-like granular or thorn-shaped dense aggregations within the proximal or distal portions of the foot processes were observed (Fig. 3i–k). Coiled bodies, which represented oligodendrocytic aggregations of hyperphosphorylated tau, were observed in the white matter (Fig. 3l). These glial 4R-tau aggregations were fundamentally argyrophilic (Fig. 3m). RD3 immunostaining was negative in most regions. Typical tufted astrocytes, astrocytic plaques, or globular glial inclusions were not observed. Argyrophilic grains were observed in the entorhinal cortex, the ambient gyrus, the subiculum, and the CA1, corresponding to stage III of Saito’s grading (Fig. 3n) [29]. Anti-phosphorylated TDP-43 immunohistochemistry revealed occasional dot-like aggregations within the neuronal cytoplasm in the hippocampus and the gray matter of the brainstem. Mislocalization of TDP-43 from the nuclei was not observed. Mild deposition of beta-amyloid was found in the frontal cortex, corresponding to Thal’s phase 1 [31] and grade A of the Consortium to Establish a Registry for Alzheimer’s Disease [24]. Anti-alpha-synuclein immunohistochemistry was negative in the brain, spinal cord, and pericardial sympathetic nerves. The hippocampal granular cells did not show immunostaining of poly-GA dipeptide, although several aggregates of 4R-tau were immunopositive for p62. Group atrophies of the muscle fibers were not observed in the intercostal, biceps, and iliopsoas muscles, the diaphragm, or the tongue. These muscle fibers did not show myogenic atrophies, internal nuclei, or ring fibers. The systemic organs showed no remarkable lesions causative of respiratory failure and death. Neuropathologic findings of patient II-2. On a coronal section of the left hemisphere, the basal ganglia and the frontotemporal cortex are not atrophied (a), whereas the subiculum shows mild atrophy (b). Severe discoloration is observed in the substantia nigra (c). The medullary reticular formation is atrophied (d, arrow). The fasciculus anterolateralis of the spinal cord shows tract degeneration, whereas the lateral column is spared (e, arrow). For comparison, photomicroscopies from a healthy control (60 years old, female) are also shown (f, g). The superficial layers of the temporal tip (h) demonstrate loss of neurons, astrogliosis, and spongiotic changes. The melanin-containing neurons are severely depleted in the pars reticulate of the substantia nigra (i). Ballooned neurons in the temporal cortices (j) and the anterior horn of spinal cord (k, arrow) are shown. d–g Klüver–Barrera staining and h–k hematoxylin–eosin staining were used. Scale bars: a–g 1 cm, h, i 100 μm, and j, k 20 μm Tau-immunopositive inclusions. Anti-AT8 immunohistochemistry reveals prominent neuronal and astrocytic aggregations in the deep layer of the cerebral cortex (a). Tau aggregation is also severe in the medullary reticular formation (b) and the anterior horn of the spinal cord (c). The intramedullary tract of the hypoglossal nerve shows intraaxonal tau aggregation (d). The tau aggregations were four-repeat tau-immunopositive (a). The neuronal inclusions comprised globose-type neurofibrillary tangles (f) and pretangles (g), and a subset of the motor neurons in the spinal anterior horn show tau inclusions with fine, fibrous configurations (h). The astrocytes demonstrate miscellaneous aggregations, including bush-like inclusions (i), and depositions within the distal (j) and proximal foot processes (k). Oligodendroglial inclusions are abundant in the cerebral white matter (l). Astrocytic inclusions are fundamentally argyrophilic (m). The hippocampal pyramidal neurons show argyrophilic grains (n). a–d, f–l Anti-AT8 immunohistochemistry, e anti-RD4 immunohistochemistry, and m, n Gallyas–Braak staining were used. Scale bars: a–d 100 μm, e, j, k 20 μm f–i, l–n 10 μm Double immunofluorescence for AT8 combined with TH, TrOH, or ChAT was performed to assess tau aggregation within the neurons of the ventrolateral medullary (VLM) nucleus, raphe nucleus, or nucleus ambiguus, respectively, of the medullary reticular formation [28]. The fluorescent signal of AT-8 was more prominently colocalized with ChAT in the nucleus ambiguus than with TH in the VLM nucleus or TrOH in the raphe nucleus (Fig. 4a–i). AT-8-immunopositive neurons occasionally contained dot-like aggregations of phosphorylated TDP-43 (Fig. 4j–l). The distributions of neuronal and glial tau aggregations are summarized in Table 1. Double immunofluorescence. Panels a–i show anti-AT8 immunohistochemistry in the medullary reticular formation. Tau aggregations are more prominent in the cholinergic neurons of the nucleus ambiguus (a–c) than in TH-immunopositive cathecolaminergic neurons in the ventrolateral medullary nucleus (d–f) or TrOH-immunopositive serotonergic neurons in the raphe nucleus (g–i). Panels j–l demonstrate occasional immunopositivity of phosphorylated TDP-43 within neurons containing AT8-immunopositive inclusions of the hypoglossal nerve. Abbreviations: ChAT, choline acetyltransferase; N. ambiguus, nucleus ambiguus; N. hypoglossal nerve, nucleus of the hypoglossal nerve; N. raphe, raphe nucleus; TH, tyrosine hydroxylase; and TrOH, tryptophan hydroxylase. Scale bars: a–c 50 μm, d–i 100 μm, and j–l 10 μm Anti-tau (T46) immunoblot of sarkosyl-insoluble fractions from the frontal cortex and striatum revealed major doublets at 64 and 68 kDa and bands at 33 and 37 kDa. The blotting bands at 33 kDa and 37 kDa were equally prominent. These blotting patterns were different from those of PSP or CBD, which would be characterized by a prominent band at either 33 kDa or 37 kDa, respectively [2] (Fig. 5a). Western blot and immune electron microscopy of aggregated tau protein. a Sarkosyl-insoluble fractions of brain tissue lysates from the frontal cortex (Fr) and caudate nucleus of our patient were subjected to western blot analysis. Patients with progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD) were also assessed for comparison. Western blot analysis using anti-T46 antibody exhibited bands prominent at 64 kDa, 68 kDa, 33 kDa, and 37 kDa. The bands at 33 kDa and 37 kDa were equally intense in our patient, whereas those at 33 kDa or 37 kDa were prominent in the patient with PSP or CBD, respectively. b, c The sarkosyl-insoluble fraction contained ribbon-like filaments with long periodicity, which were immunopositive for AT8 (b) and an anti-Tau C antibodies (c). Scale bars = 100 nm, original magnification × 60,000 Tau fibrils, which were isolated from the sarkosyl-insoluble fraction, had diameters of 15–20 μm and exhibited long periodicity and immunoreactivity to anti-AT8 and anti-tau C terminal antibodies (Fig. 5b, c). There were no known pathogenic or rare variants in the causative genes related to Perry syndrome, FTD, familial Parkinson’s disease, parkinsonism, or Alzheimer’s disease.


Outcomes of western blot

Outcomes of immunoelectron microscopy
Outcomes of genetic analysis
Discussion and conclusions
The patient died of respiratory failure 14 months after the presentation of parkinsonism. A postmortem study revealed neuronal and glial aggregation of 4R-tau. Tau aggregations and neuronal loss were prominent in the medial temporal lobes, the temporal tip, the striatum, the substantia nigra, the tegmentum of the midbrain, and the medullary reticular formation. Astrocytic inclusions comprised granular or dense aggregations of the proximal or distal portion of the foot processes; the morphological features were similar to those of the bush-like or thorn-shaped astrocytes in ARTAG [19]. Oligodendroglia showed the appearance of coiled bodies but not globular glial inclusions [17]. Western blot analysis of the sarkosyl-insoluble fraction revealed a low molecular band equally prominent at 33 and 37 kDa [2]. On electron microscopy, the aggregated tau fibrils were relatively straight with long periodicity, which was similar to those in PSP and CBD [3]. These clinical findings, morphological and biochemical findings of tau aggregations, and distributions of tau aggregations and neuronal loss could not be classified into known 4R-tau-related neurodegenerative disorders, including PSP, CBD, GGT, ARTAG, and AGD.
Her sibling also showed rapidly progressive parkinsonism and sudden death. Unfortunately, a postmortem study was not undertaken for him, and it was unclear if he also had 4R-tau aggregation. However, the familial clustering of neurological manifestations suggests a genetic background. We did not identify pathologic mutations in MAPT or DCTN-1, which are causative of familial tauopathy and Perry syndrome, respectively [6, 12]. The other genetic variants related to parkinsonism and cognitive disorders were also not detected either. We could not evaluate the possibility of a novel gene by whole genome sequencing or linkage analysis due to the small number of family members. We expect further investigation to find any novel gene variants, which characterize this unclassifiable 4R-tauopathy with clinically aggressive manifestations and the chance of familial history.
Sporadic or familial patients with unclassifiable 4R-tau aggregations have occasionally been reported [4, 7, 15, 18]. Although gene variants in MAPT or FUS have been suggested, the pathogenesis remains unclear [7, 18]. FTD, parkinsonism or both have been observed in previous reports. Important clinical features of our patient were nonmotor symptoms of progressive respiratory failure with hypercapnia, weight loss, and sleep disturbance. The postmortem assay revealed prominent aggregations of tau in the medullary reticular formation, particularly in the nucleus ambiguus. The region is known as a part of the ventral respiratory group and as a parasympathetic center. Its pathologic involvement might contribute to those nonmotor symptoms, as presumed for the autonomic dysfunctions of Perry syndrome [26]. Another finding of interest was severe tau aggregations in the lower motor neurons of the spinal cord. A subset of neuronal inclusions showed AT-8-immunopositive fibrous configurations that differed from pretangles or globose-type tangles. The skeletal muscles did not show neurogenic atrophy, and the lower motor neurons were not depleted; it remains unclear whether the tau aggregations of the lower motor neurons impaired muscle strength and ventilation. Prominent tau aggregation within the lower motor neurons has also been reported among patients with GGT, and those patients sometimes demonstrate motor neuron disease-like muscle weakness [8]. The limitation of this study is lack of comprehensive neurophysiological assessments including electromyography, respiratory monitoring, and polysomnography. Detailed assessments of the respiratory and autonomic systems could further reveal the pathomechanism of the various nonmotor symptoms of the patient.
Argyrophilic grains may contribute to atrophy and the presence of ballooned neurons in the medial temporal structures of our patient. Several studies have demonstrated patients with occasional diffuse AGD (DAGD) that involves not only the medial temporal lobes but also the neocortices, limbic cortices, and the brainstem [14, 22]. FTD-like behavioral disorders, mood disorders, or mild parkinsonism have been described in these patients [14, 22]. A recent study revealed that extensions of granular or fuzzy astrocytes were positively correlated with progression of AGD [23]. However, neuronal loss and neuronal and astrocytic tau aggregations in our patient were clearly broader than the presence of argyrophilic grains. The western blot of our patient exhibited a low molecular band equally prominent at 33 and 37 kDa, whereas that of AGD patients would show quite weak bands in those molecular weights [23]. We consider that the clinicopathological phenotypes of our patient cannot be explained by AGD alone. A prominent tau aggregation in the brainstem or the spinal cord could be indicative of IgLON5-related tauopathy. However, this disease entity usually displays a combination of 3R and 4R-tauopathy [9]. Myotonic dystrophy is also associated with tauopathies but usually demonstrates 3R tauopathy or a combination of 3R and 4R tauopathies [5, 20, 25, 32]. In addition, skeletal muscle of our patient did not have pathological features of myotonic dystrophy. Those facts differentiate our patient from IgLON5-related or myotonic dystrophy-related tauopathy, although assessments of HLA haplotypes or genetic analyses of DMPK and CNBP genes were not available. Neuronal aggregations of phosphorylated TDP-43 were influent but always colocalized with tau aggregations in our patient. The results of anti-p62 and anti-dipeptide immunohistochemistry and a C9orf72 gene analysis suggest that TDP-43 aggregation in our patient might be induced by 4R-tau aggregation [16] rather than primary TDP-43 proteinopathy.
In this report, we described a patient who was characterized by parkinsonism and progressive respiratory failure in association with unclassified 4R-tau aggregation. Our results further extend the clinical and neuropathologic spectra of 4R-tauopathy.
Availability of data and materials
All clinical data and material from the pathology are available from Mari Yoshida (myoshida@aichi-med-u.ac.jp).
Abbreviations
- 4R-tau:
-
four-repeat tau
- 3R-tau:
-
three-repeat tau
- AGD:
-
argyrophilic grain dementia
- ARTAG:
-
aging-related tau astrogliopathy
- CA:
-
cornu ammonis
- CBD:
-
corticobasal degeneration
- ChAT:
-
choline acetyltransferase
- DCTN-1:
-
dynactin subunit 1
- DNA:
-
deoxyribonucleic acid
- FTD:
-
frontotemporal dementia
- GGT:
-
globular glial tauopathy
- MAPT:
-
microtubule-associated protein tau
- PiD:
-
Pick’s disease
- PSEN-1:
-
presenilin-1
- PSP:
-
progressive supranuclear palsy
- TDP-43:
-
TAR DNA binding protein 43 kDa
- TH:
-
tyrosine hydroxylase
- TrOH:
-
tryptophan hydroxylase
- VLM nucleus:
-
ventrolateral medullary nucleus
References
Andreadis A, Brown WM, Kosik KS (1992) Structure and novel exons of the human tau gene. Biochemistry 31:10626–10633
Arai T, Ikeda K, Akiyama H, Nonaka T, Hasegawa M, Ishiguro K et al (2004) Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 55:72–79
Arima K (2006) Ultrastructural characteristics of tau filaments in tauopathies: immuno-electron microscopic demonstration of tau filaments in tauopathies. Neuropathology. 26:475–483
Bigio EH, Lipton AM, Yen SH, Hutton ML, Baker M, Nacharaju P et al (2001) Frontal lobe dementia with novel tauopathy: sporadic multiple system tauopathy with dementia. J Neuropathol Exp Neurol 60:328–341
Delacourte A, Buée L (2000) Tau pathology: a marker of neurodegenerative disorders. Curr Opin Neurol 13:371–376
Farrer MJ, Hulihan MM, Kachergus JM, Dächsel JC, Stoessl AJ, Grantier LL et al (2009) DCTN1 mutations in Perry syndrome. Nat Genet 41:163–165
Ferrer I, Legati A, García-Monco JC, Gomez-Beldarrain M, Carmona M, Blanco R et al (2015) Familial behavioral variant frontotemporal dementia associated with astrocyte-predominant tauopathy. J Neuropathol Exp Neurol 74:370–379
Fu YJ, Nishihira Y, Kuroda S, Toyoshima Y, Ishihara T, Shinozaki M et al (2010) Sporadic four-repeat tauopathy with frontotemporal lobar degeneration, parkinsonism, and motor neuron disease: a distinct clinicopathological and biochemical disease entity. Acta Neuropathol 120:21–32
Gelpi E, Höftberger R, Graus F, Ling H, Holton JL, Dawson T et al (2016) Neuropathological criteria of anti-IgLON5-related tauopathy. Acta Neuropathol 132:531–543
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA (1989) Cloning and sequencing of cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J 8:393–399
Hayashida A, Li Y, Yoshino H, Daida K, Ikeda A, Ogaki K et al (2020) The identified clinical features of Parkinson’s disease in homo-, heterozygous and digenic variants of PINK1. Neurobiol Aging. https://doi.org/10.1016/j.neurobiolaging.2020.06.017
Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H et al (1998) Association of missense and 5′-splice mutations in tau with the inherited FTDP-17. Nature 393:702–705
Ikeda A, Shimada H, Nishioka K, Takanashi M, Hayashida A, Li Y et al (2019) Clinical heterogeneity of frontotemporal dementia and parkinsonism linked to chromosome 17 caused by MAPT N279K mutation in relation to tau positron emission tomography features. Mov Disord 34:568–574
Ishihara K, Araki S, Ihori N, Shiota J, Kawamura M, Yoshida M et al (2005) Argyrophilic grain disease presenting with frontotemporal dementia: a neuropsychological and pathological study of an autopsied case with presenile onset. Neuropathology 25:165–170
Iwasaki Y, Mori K, Ito M, Tatsumi S, Mimuro M, Kuwano R et al (2016) An autopsied case of unclassifiable sporadic four-repeat tauopathy presenting with parkinsonism and speech disturbances. Neuropathology 36:295–304
Koga S, Sanchez-Contreras M, Josephs KA, Uitti RJ, Graff-Radford N, van Gerpen JA et al (2017) Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord 32:246–255
Kovacs GG, Majtenyi K, Spina S, Murrell JR, Gelpi E, Hoftberger R et al (2008) White matter tauopathy with globular glial inclusions: a distinct sporadic frontotemporal lobar degeneration. J Neuropathol Exp Neurol 67:963–975
Kovacs GG, Wöhrer A, Ströbel T, Botond G, Attems J, Budka H (2011) Unclassifiable tauopathy associated with an A152T variation in MAPT exon 7. Clin Neuropathol 30:3–10
Kovacs GG, **e SX, Lee EB, Robinson JL, Caswell C, Irwin DJ et al (2017) Multisite assessment of aging-related tau astrogliopathy (ARTAG). J Neuropathol Exp Neurol 76:605–619
Kuru S, Sakai M, Yoshida M, Mimuro M, Hashizume Y (2013) Neurofibrillary tangles in the peripheral sympathetic ganglia of patients with myotonic dystrophy. Neuropathol Appl Neurobiol 39:585–588
Mann DMA, Snowden JS (2017) Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathol 27:723–736
Maurage CA, Sergeant N, Schraen-Maschke S, Lebert F, Ruchoux MM, Sablonnière B et al (2003) Diffuse form of argyrophilic grain disease: a new variant of four-repeat tauopathy different from limbic argyrophilic grain disease. Acta Neuropathol 106:575–583
Miki T, Yokota O, Haraguchi T, Ishizu H, Hasegawa M, Ishihara T et al (2020) Factors associated with development and distribution of granular/fuzzy astrocytes in neurodegenerative diseases. Brain Pathol 30:811–830
Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The consortium to establish a registry for alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Mizuno Y, Maeda N, Hamasaki H, Arahata H, Sasagasako N, Honda H et al (2018) Four-repeat tau dominant pathology in a congenital myotonic dystrophy type 1 patient with mental retardation. Brain Pathol 3:431–433
Ohshima S, Tsuboi Y, Yamamoto A, Kawakami M, Farrer MJ, Kira J, Shii H (2010) Autonomic failures in Perry syndrome with DCTN1 mutation. Parkinsonism Relat Disord 16:612–614
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72:257–268
Riku Y, Watanabe H, Mimuro M, Iwasaki Y, Ito M, Katsuno M et al (2017) Non-motor multiple system atrophy associated with sudden death: pathological observations of autonomic nuclei. J Neurol 264:2249–2257
Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y et al (2004) Staging of argyrophilic grains: an age-associated tauopathy. J Neuropathol Exp Neurol 63:911–918
Taniguchi-Watanabe S, Arai T, Kametani F, Nonaka T, Masuda-Suzukake M, Tarutani A et al (2016) Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol 131:267–280
Thal DR, Rüb U, Orantes M, Braak H (2002) Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 58:1791–1800
Vermersch P, Sergeant N, Ruchoux MM, Hofmann-radvanyi HH, Wattez A, Petit H et al (1996) Specific tau variants in the brains of patients with myotonic dystrophy. Neurology 3:711–717
Ion AmpliSeq Designer. https://www.ampliseq.com
Acknowledgements
We appreciate the patient’s family for providing tissue and genetic materials.
Funding
The authors are very grateful for these grants: AMED-CREST (Japan Agency of Medical Research and Development), Practical Research Project for Rare/Intractable Diseases from AMED, 15ek0109029s0202 (N.H.); AMED under grant numbers JP18dm0107105, 19dm0107105, and JP16kk0205009, and Grants-in-Aid from the Research Committee of CNS Degenerative Diseases, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan (M.Y.).
Author information
Authors and Affiliations
Contributions
Autopsy (A.A., M.H., and Y.I.), clinical analysis (M.N., Y.W., Y.A., A.T., K.H., A.Y., K.S., and M.T.), pathologic analysis (Y.R. and M.Y.), biochemical analysis (M.H.), genetic analysis (K.N., Y.L., H.Y., and N.H.), drafting manuscript (M.N.), and manuscript supervision (Y.R. and M.Y.). All authors read and approved the final manuscript
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study is in compliance with the ethical committee for medical research of Aichi Medical University (committee acceptance number: AKBRC15-017).
Consent for publication
All authors agree with the submission of this manuscript to the designated journal.
Competing interests
We have no conflicts of interest to report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nakano, M., Riku, Y., Nishioka, K. et al. Unclassified four-repeat tauopathy associated with familial parkinsonism and progressive respiratory failure. acta neuropathol commun 8, 148 (2020). https://doi.org/10.1186/s40478-020-01025-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40478-020-01025-1