
Abstract
Background
Metabolic associated steatohepatitis (MASH) is metabolic disease that may progress to cirrhosis and hepatocellular carcinoma. Mouse models of diet-induced MASH, which is characterized by the high levels of fats, sugars, and cholesterol in diets, are commonly used in research. However, mouse models accurately reflecting the progression of MASH in humans remain to be established. Studies have explored the potential use of serological metabolites as biomarkers of MASH severity in relation to human MASH.
Methods
We performed a comparative analysis of three mouse models of diet-induced MASH in terms of phenotypic and metabolomic characteristics; MASH was induced using different diets: a high-fat diet; a Western diet; and a high-fat, high-cholesterol diet. Liver cirrhosis was diagnosed using standard clinical approaches (e.g., METAVIR score, hyaluronan level, and collagen deposition level). Mouse serum samples were subjected to nuclear magnetic resonance spectroscopy–based metabolomic profiling followed by bioinformatic analyses. Metabolomic analysis of a retrospective cohort of patients with hepatocellular carcinoma was performed; the corresponding cirrhosis scores were also evaluated.
Results
Using clinically relevant quantitative diagnostic methods, the severity of MASH was evaluated. Regarding metabolomics, the number of lipoprotein metabolites increased with both diet and MASH progression. Notably, the levels of very low-density lipoprotein (VLDL) and low-density lipoprotein (LDL) significantly increased with fibrosis progression. During the development of diet-induced MASH in mice, the strongest upregulation of expression was noted for VLDL receptor. Metabolomic analysis of a retrospective cohort of patients with cirrhosis indicated lipoproteins (e.g., VLDL and LDL) as predominant biomarkers of cirrhosis.
Conclusions
Our findings provide insight into the pathophysiology and metabolomics of experimental MASH and its relevance to human MASH. The observed upregulation of lipoprotein expression reveals a feedforward mechanism for MASH development that may be targeted for the development of noninvasive diagnosis.
Similar content being viewed by others


Background
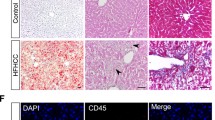
Metabolic dysfunction-associated steatotic liver disease (MASLD) is formerly known as nonalcoholic fatty liver disease (NAFLD) and is a major cause of chronic liver disease worldwide [1,2,3,4]. MASLD can develop as hepatic steatosis or severe conditions involving inflammation, such as metabolic dysfunction-associated steatohepatitis (MASH), liver fibrosis, cirrhosis, hepatocellular carcinoma (HCC), and liver failure [3, 4]. MASH is characterized by hepatic inflammation, hepatocyte ballooning, and intrahepatic fat accumulation [5, 6]. Patients with MASH are at a higher risk of liver cancer, liver failure, and cardiovascular disease than are healthy individuals [2, 5]. Key risk factors for MASH include diabetes, obesity, age, ethnicity, sex, and genetic polymorphisms, which can also affect MASH progression [7, 6B). Pathological analysis (Fig. 2) revealed that although both WD and HFC induced MASH, HFC induced a more severe condition—liver fibrosis. Therefore, we further compared the WD group (mild fibrosis) with the HFC group (severe fibrosis) to explore fibrosis-specific metabolites. A total of 17 metabolites were found to be associated with severe liver fibrosis (Fig. 6C). VLDLs and LDLs accounted for 35% and 47%, respectively, of the aforementioned metabolites; both VLDLs and LDLs were found to be predominant in the liver of mice with severe MASH (Fig. 6C). The 17 markers included the large VLDLs V1CH, V1PL, V2CH, V2TG, and V4CH; the small LDLs L5PN, L5CH, L5PL, and L5AB; and the HDL HDTG (Fig. 6C). Taken together, the results indicate VLDL and LDL are involved in the development of MASLD/MASH and induce severe fibrosis. These findings elucidate both the roles of VLDL and LDL as biomarkers of severe MASH and the pathophysiological changes that occur during the progression of MASH.

Expression levels of very low–density lipoprotein and low-density lipoprotein are strongly correlated with the severity of metabolic dysfunction-associated steatohepatitis/fibrosis. A Correlation between serological metabolites and collagen scores. Significant lipoproteins were selected on the basis of a p value of < 0.05 and a correlation coefficient of > 0.3. B Integrated analysis of significant metabolites and metabolites strongly associated with fibrosis. Blue indicates significant metabolites, whereas yellow indicates metabolites strongly correlated with fibrosis. C Shortened list of 17 significant metabolites identified from the comparison between HFC and WD groups (Table on the left). The pie chart presents the proportions of the 17 significant metabolites
Upregulation of VLDLR expression in mouse models of diet-induced MASH
The liver is the most prominent contributor of lipoproteins because this organ is responsible for both the production and recycling of lipoproteins [47]. Lipoprotein receptors are crucial for systemic lipid metabolism. The expression of lipoprotein receptors, such as VLDLR, LDL receptor (LDLR), and HDL receptor (SR-B1), in normal organs has been studied in humans and mice [47, 48]. VLDL is believed to be produced only by the liver; VLDLR is expressed in the periphery of but not within the liver [49]. In our study, the expression of both LDL and VLDL was upregulated in mouse models of severe MASH, which prompted us to investigate the receptors of these lipoproteins in diseased liver tissues. The expression and distribution of VLDLR, LDLR, and SR-B1 were evaluated through immunohistochemical analysis (Fig. 7A) and was quantified using ImageJ (Fig. 7B). The expression of VLDLR was not similar between the diet- or feeding time–based groups, with the exception of the HCF group, which was fed for 32 weeks (Fig. 7B). After 16 weeks of feeding, the expression of LDLR was markedly downregulated in the experimental groups compared with that in the control group; nonetheless, the expression was gradually restored in the WD and HFC groups after 32 weeks of feeding (Fig. 7B). Notably, the expression of SR-B1 remained high and did not change with diet (Fig. 7B). In summary, the expression of VLDLR is considerably upregulated in severe liver fibrosis. The findings of increases in the levels of serological VLDL and LDL and the upregulation of VLDLR expression in the severe MASH of this study indicate a feedforward mechanism for lipid deposition.

Expression levels of lipoprotein receptors in mouse models of diet-induced MASH. A Results of immunohistochemical staining performed to measure the expression levels of very low–density lipoprotein receptor, low-density lipoprotein receptor, and high-density lipoprotein receptor (SR-B1) in the livers of mice with diet-induced MASH. The brown indicates receptor expression. B Receptor expression levels were quantified using ImageJ and analyzed using GraphPad Prism (version 8). The white, black, blue, and red dots indicate NC, HFD, WD, and HFC, respectively. Statistical significance: *p < 0.05, compared with NC. NC: normal chow; HFC: high-fat diet; WD: Western diet; HFC: high-fat, high-cholesterol; and MASH: metabolic dysfunction-associated steatohepatitis
VLDLs serve as the biomarkers of liver fibrosis/cirrhosis in humans
The expression of large VLDLs and VLDLR are upregulated in mouse models of diet-induced MASH with severe fibrosis (Figs. 6C and 7). We analyzed the clinical specimens of a retrospective cohort of patients with liver fibrosis/cirrhosis to identify the correlation between serological metabolites and clinical features. The demographic characteristics of our cohort are summarized in Supplementary Table 5. On the basis of their METAVIR scores, the patients were stratified into mild and severe disease groups. A comparison of the metabolome and differential expression of relevant genes were performed. The results revealed significant increases in the levels of the following metabolites (very large VLDLs) in patients with severe fibrosis: XXL_VLDL_CE, XXL_VLDL_C, L_VLDL_CE, and L_VLDL_C (Fig. 8A). Human and experimental (mouse) MASH diseases were similar in terms of the upregulation of the expression of very large VLDLs. In summary, the severity of diet-induced MASH in mouse models can be evaluated to align with clinical diagnostic methods. Metabolomic profiling revealed a likely mechanism of VLDL recycling through VLDLR, which may be involved the pathogenesis of liver fibrosis/cirrhosis.

Very low–density lipoprotein and low-density lipoprotein as key biomarkers of fibrosis/cirrhosis in humans. A Significant metabolites identified through the differential gene expression analysis of a retrospective cohort of patients with fibrosis/cirrhosis. Log2 fold changes were calculated by comparing the FIB4 scores of patients with advanced disease (F3 or F4) with those of patients with no or mild fibrosis (F0–F2). Significant metabolites associated with cirrhosis. Log2 fold changes were determined by comparing patients with cirrhosis with those without cirrhosis. B Changes in lipoproteins during the progression of MASH/fibrosis in mice
Discussion
In this study, three commonly employed experimental diet–induced MASH models were used to evaluate MASH severity by using clinically relevant diagnostic methods. The serological metabolites associated with MASH severity was identified. The roles of the lipoprotein–receptor axes in the pathogenesis of diet-induced MASH in mice were investigated.
Importance of mouse models of diet-induced MASH in clinical diagnosis
MASH is diagnosed on the basis of histopathological features, such as steatosis, hepatocyte ballooning, and lobular inflammation. Although fibrosis is not a histopathological feature of MASH, it can be used to predict the risk of mortality. The METAVIR scoring system is a commonly used tool for diagnosing fibrosis. The pathogenesis and progression of MASH is complex and involve cellular heterogeneity and alterations in the humoral matrix. Histopathological analysis is important in MASH diagnosis. Rodent models of experimental MASLD/MASH/fibrosis/cirrhosis can be established through diet, genetic modifications, toxin treatment, and a combination of different methods [11, 15, 50]. Few studies have been conducted to systemically compare diet-induced MASLD/MASH models, evaluate their importance in clinical diagnosis, and identify metabolite biomarkers. The present study was conducted to obtain valuable insight into various decompensated liver diseases, such as MASH, fibrosis, and cirrhosis. Inbred rodents are widely used for studying MASLD/MASH from a genetic perspective [51]. The present study was conducted using C57BL/6 mice, which are commonly used in transgenic animal studies.
Importance of noninvasive metabolomic tools in MASH diagnosis
The METAVIR scoring system is an invasive tool that involves liver biopsy, which involves the risks of major vein rupture and internal bleeding. Serological biomarkers, such as FIB4, can serve as noninvasive tools for disease diagnosis. However, the precision of clinical diagnosis performed on the basis of FIB4 is low (receiver operating characteristic curve score, approximately 70%) [52, 53]. In our study, high-throughput metabolomic profiling was performed with NMR spectroscopy; the results revealed a correlation between the phenotypic and metabolomic characteristics of mouse models of MASH. In addition, a novel biomarker of steatohepatitis was identified.
Regarding the relevance of experimental MASH to human MASH, elevated levels of VLDL, VLDL-cholesterol, and LDL-cholesterol can serve as the biomarkers of the progression of hepatic steatosis to MASH (Fig. 8A). High serum levels of total lipid and cholesterol (VLDL and LDL) are associated with intrahepatic cholesterol accumulation and hepatocyte injury in MASH [54, 55]. The similarity between experimental diet-induced MASH and human MASH in terms of lipoprotein metabolites indicated lipoprotein analysis may be valuable for clinical diagnosis. Therefore, the significance of our study lies in its identification of an association of large VLDLs and LDLs with the progression of MASH in animal models and patients (Fig. 8B).
Most studies conducted using animal models of MASH have explored lipid, glucose, and protein metabolites in the liver. Our findings reveal that the levels of lipoproteins increased with the severity of MASH. Through NMR spectroscopy–based metabolomic profiling, both VLDL and LDL were simultaneously explored in mice and humans. The consistency between the experimental MASH and human MASH in terms of metabolite biomarkers indicated that similarities were present in the pathophysiological alternations between the mouse and human, and that NMR spectroscopy–based metabolomic profiling can be valuable for research and clinical diagnosis.
Lipoproteins as the predominant biomarkers of diet-induced MASH in mice
Serum-insoluble lipids circulate in the bloodstream as lipoproteins, which are macromolecular complexes of free cholesterol, cholesterol esters, triglycerides, phospholipids, and apolipoproteins [56]. The liver is the primary site for the synthesis of LDL (16–30 nm) and VLDL (30–80 nm), which carry lipids and ApoB100 to tissue. An essential lipoprotein for the collection and transportation of extra serum cholesterol is HDL (8–16 nm; ApoA). Structurally, VLDL comprises a triglyceride-enriched core surrounded by a monolayer of phospholipids and incorporated proteins (e.g., ApoB-100), which facilitates the delivery and uptake of VLDL. Notably, most triglycerides incorporated into VLDL are derived from exogenous lipids and not synthesized through de novo lipogenesis [57, 58]. The impaired synthesis of ApoB-100 in patients with MASH may be associated with increased free fatty acid level, disrupted redox balance, hyperinsulinemia, and reduced gene expression, all of which hinder ApoB-100 synthesis and VLDL assembly, thus resulting in intrahepatic lipid accumulation [59]. Additionally, VLDL particles can be converted into LDL particles through hydrolyzation of triglycerides by LPL in the bloodstream [56].
In patients with MASLD, the expression of VLDL in the liver is upregulated, leading to increased levels of triglycerides. In addition, the clearance of LDL is reduced, which accelerates the development of atherosclerosis and cardiovascular disease. Excessive lipid storage in the liver promotes the secretion of VLDL and thus dyslipidemia [60]. Furthermore, an increase in the level of oxidized LDL occurs, which induces systemic inflammation [60].
The increase in the mean size of VLDL in patients with MASH and the reduction in the level of small VLDL in patients with liver fibrosis reflect changes in the number and state of hepatocytes resulting from such diseases [61]. Hepatocytes with increased levels of intracellular lipid can serve as the source of large VLDLs. MASH driven by insulin resistance and an increase in the intrahepatic lipid pool may increase the numbers of large VLDLs and the mean size of VLDLs [62,63,64].
Upregulation of VLDL/VLDLR expression indicates a positive feedforward mechanism for hepatic lipid accumulation
In adipose tissues, VLDLR is regulated by peroxisome proliferator–activated receptor [65]. Free cholesterol and fatty acid can promote stress response, inflammation, apoptosis, and fibrosis in the liver [66]. Large VLDLs induce the accumulation of triglycerides in macrophages and exhibit a higher affinity for VLDLR binding than do small VLDLs. Disease states may influence VLDL properties. Hepatic secretion of VLDL is impaired in patients with ApoB mutations, which often leads to fatty liver disease because of the excessive intrahepatic accumulation of fat. The extent of steatosis is associated with the size and number of VLDLs in the patient population [67]. Patients with hepatic steatosis and insulin resistance have increased levels of circulating ApoC-III, which is a strong inhibitor of LPL. After the LPL-mediated hydrolysis of triglycerides, lipoprotein remnants are removed through receptor-mediated pathways, primarily those operated in the liver. Obesity and insulin resistance contribute to reduced LDL clearance by reducing the activities of LDLR and LDLR-related protein 1, among others [68]. The intrahepatic accumulation of LDL due to reduced receptor-mediated uptake may directly inhibit LPL, resulting in a feedforward mechanism that drives lipid deposition and MASH development [69,70,71].
The dysregulation of lipid homeostasis in hepatocytes leads to the generation of toxic lipids that impair organelle functions, promoting inflammation, hepatocellular damage, and apoptosis [69]. In patients with MASH, the uptake of circulating lipids, particularly free fatty acids and lipoproteins, by the liver is higher [72]. Because the hepatic secretion of lipoprotein is higher in patients with MASLD, the deposition of fat in hepatocytes disrupts lipid homeostasis in these cells [72]. Further studies are required to evaluate the quantity and quality of changes in lipid metabolites during the pathogenesis of MASH.
Conclusions
Our findings provide key insight into the pathophysiology and serological metabolomics of experimental diet-induced MASH in relation to human MASH. The finding of an upregulation of lipoprotein expression indicates a feedforward mechanism underlies MASH development, and this mechanism may be targeted for the development of noninvasive diagnostic strategies.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
References
Younossi ZM. Non-alcoholic fatty liver disease - A global public health perspective. J Hepatol. 2019;70(3):531–44.
Sheka AC, et al. Nonalcoholic Steatohepatitis: A Review. JAMA. 2020;323(12):1175–83.
Chen YY, Yeh MM. Non-alcoholic fatty liver disease: A review with clinical and pathological correlation. J Formos Med Assoc. 2021;120(1 Pt 1):68–77.
Fon Tacer KD. Rozman, Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. J Lipids. 2011;2011:783976.
Peng C, et al. Non-Alcoholic Steatohepatitis: A Review of Its Mechanism Models and Medical Treatments. Front Pharmacol. 2020;11:603926.
Younossi Z, et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol. 2018;15(1):11–20.
Sulaiman SA, Dorairaj V, Adrus MNH. Genetic Polymorphisms and Diversity in Nonalcoholic Fatty Liver Disease (NAFLD): A Mini Review. Biomedicines. 2022;11(1):106.
**a MF, Bian H, Gao X. NAFLD and Diabetes: Two Sides of the Same Coin? Rationale for Gene-Based Personalized NAFLD Treatment. Front Pharmacol. 2019;10:877.
Haas JT, Francque S, Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu Rev Physiol. 2016;78:181–205.
Recena Aydos L, et al. Nonalcoholic Fatty Liver Disease Induced by High-Fat Diet in C57bl/6 Models. Nutrients. 2019;11(12):3067.
Hansen HH, et al. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22(11):1707–18.
Radhakrishnan S, Ke JY, Pellizzon MA. Targeted Nutrient Modifications in Purified Diets Differentially Affect Nonalcoholic Fatty Liver Disease and Metabolic Disease Development in Rodent Models. Curr Dev Nutr. 2020;4(6):nzaa078.
Lee SJ, et al. Proteomic analysis of mice fed methionine and choline deficient diet reveals marker proteins associated with steatohepatitis. PLoS One. 2015;10(4):e0120577.
Caballero F, et al. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J Biol Chem. 2010;285(24):18528–36.
Jiang M, et al. Pathogenesis of and major animal models used for nonalcoholic fatty liver disease. J Int Med Res. 2019;47(4):1453–66.
Lau JK, Zhang X, Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J Pathol. 2017;241(1):36–44.
Jensen T, et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J Hepatol. 2018;68(5):1063–75.
Ioannou GN. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol Metab. 2016;27(2):84–95.
Subramanian S, et al. Dietary cholesterol exacerbates hepatic steatosis and inflammation in obese LDL receptor-deficient mice. J Lipid Res. 2011;52(9):1626–35.
Savard C, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology. 2013;57(1):81–92.
Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol. 2018;68(2):230–7.
Kohli R, et al. High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology. 2010;52(3):934–44.
Charlton M, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G825–34.
Lee JS, et al. Histologic and Metabolic Derangement in High-Fat, High-Fructose, and Combination Diet Animal Models. Sci World J. 2015;2015:306326.
Tri Reagent for RNA Isolation from tissues cells. Sigma-Aldrich Co. LLC., 2021.
PrimeScriptTM RT reagent Kit (Perfect Real Time). Takara Bio Inc, 2022.
Van De Vlekkert D, Machado E, d’Azzo A. Analysis of Generalized Fibrosis in Mouse Tissue Sections with Masson’s Trichrome Staining. Bio Protoc. 2020;10(10):e3629.
Li C, Li R, Zhang W. Progress in non-invasive detection of liver fibrosis. Cancer Biol Med. 2018;15(2):124–36.
**e C, et al. Comparison of serological assessments in the diagnosis of liver fibrosis in bile duct ligation mice. Exp Biol Med (Maywood). 2017;242(14):1398–404.
Yeh CL, et al. Shear-wave elasticity imaging of a liver fibrosis mouse model using high-frequency ultrasound. IEEE Trans Ultrason Ferroelectr Freq Control. 2015;62(7):1295–307.
Xu L, et al. Remdesivir Inhibits Tubulointerstitial Fibrosis in Obstructed Kidneys. Front Pharmacol. 2021;12:626510.
Wurtz P, et al. Quantitative Serum Nuclear Magnetic Resonance Metabolomics in Large-Scale Epidemiology: A Primer on -Omic Technologies. Am J Epidemiol. 2017;186(9):1084–96.
Yap BW, Sim CH. Comparisons of various types of normality tests. J Stat Comput Simul. 2011;81(12):2141–55.
Chen Y, Li EM, Xu LY. Guide to Metabolomics Analysis: A Bioinformatics Workflow. Metabolites. 2022;12(4):357.
Pasikanti KK, et al. Noninvasive urinary metabonomic diagnosis of human bladder cancer. J Proteome Res. 2010;9(6):2988–95.
Benton PH, et al. An Interactive Cluster Heat Map to Visualize and Explore Multidimensional Metabolomic Data. Metabolomics. 2015;11(4):1029–34.
Draisma HH, et al. Hierarchical clustering analysis of blood plasma lipidomics profiles from mono- and dizygotic twin families. Eur J Hum Genet. 2013;21(1):95–101.
Vinaixa M, et al. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites. 2012;2(4):775–95.
Pan YY, et al. Visualization of statistically processed LC-MS-based metabolomics data for identifying significant features in a multiple-group comparison. Chemom Intell Lab Syst. 2021;210:104271.
Razali NM, Wah YB. Power comparisons of Shapiro-Wilk, Kolmogorov-Smirnov, Lilliefors and Anderson-Darling tests. J Stat Model Anal. 2011;2(1):21–33.
Kim HY. Statistical notes for clinical researchers: Nonparametric statistical methods: 2. Nonparametric methods for comparing three or more groups and repeated measures. Restor Dent Endod. 2014;39(4):329–32.
Wang WW, et al. Altered fecal microbiome and metabolome in adult patients with non-cystic fibrosis bronchiectasis. Respir Res. 2022;23(1):317.
Chen SY, Feng Z, Yi X. A general introduction to adjustment for multiple comparisons. J Thorac Dis. 2017;9(6):1725–9.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Ser B (Methodol). 1995;57(1):289–300.
Qi Z, Voit EO. Strategies for Comparing Metabolic Profiles: Implications for the Inference of Biochemical Mechanisms from Metabolomics Data. IEEE/ACM Trans Comput Biol Bioinform. 2017;14(6):1434–45.
Rossi E, et al. Assessing liver fibrosis with serum marker models. Clin Biochem Rev. 2007;28(1):3–10.
Feingold KR. Lipid and Lipoprotein Metabolism. Endocrinol Metab Clin North Am. 2022;51(3):437–58.
Ramasamy I. Recent advances in physiological lipoprotein metabolism. Clin Chem Lab Med. 2014;52(12):1695–727.
Webb JC, et al. Characterization and tissue-specific expression of the human “very low density lipoprotein (VLDL) receptor” mRNA. Hum Mol Genet. 1994;3(4):531–7.
Flessa CM, et al. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int J Mol Sci. 2022;23(24):15791.
Jahn D, et al. Animal models of NAFLD from a hepatologist’s point of view. Biochim Biophys Acta Mol Basis Dis. 2019;1865(5):943–53.
Park SH, Goo JM, Jo CH. Receiver operating characteristic (ROC) curve: practical review for radiologists. Korean J Radiol. 2004;5(1):11–8.
Park H, et al. Reappraisal of fibrosis-4 index and non-alcoholic fatty liver disease fibrosis score for advanced fibrosis in average-risk population. Front Med (Lausanne). 2022;9:1024836.
Mannisto VT, et al. Lipoprotein subclass metabolism in nonalcoholic steatohepatitis. J Lipid Res. 2014;55(12):2676–84.
Corey KE, et al. Non-high-density lipoprotein cholesterol as a biomarker for nonalcoholic steatohepatitis. Clin Gastroenterol Hepatol. 2012;10(6):651–6.
Smith LC, Pownall HJ, Gotto AM Jr. The plasma lipoproteins: structure and metabolism. Annu Rev Biochem. 1978;47:751–7.
Donnelly KL, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–51.
Fabbrini E, et al. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):424–31.
Perla FM, et al. The Role of Lipid and Lipoprotein Metabolism in Non-Alcoholic Fatty Liver Disease. Children (Basel). 2017;4(6):46.
Heeren J, Scheja L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol Metab. 2021;50:101238.
Jiang ZG, et al. Steatohepatitis and liver fibrosis are predicted by the characteristics of very low density lipoprotein in nonalcoholic fatty liver disease. Liver Int. 2016;36(8):1213–20.
Mora S, et al. Lipoprotein particle size and concentration by nuclear magnetic resonance and incident type 2 diabetes in women. Diabetes. 2010;59(5):1153–60.
Chan DC, et al. Nonalcoholic fatty liver disease as the transducer of hepatic oversecretion of very-low-density lipoprotein-apolipoprotein B-100 in obesity. Arterioscler Thromb Vasc Biol. 2010;30(5):1043–50.
Adiels M, et al. Acute suppression of VLDL1 secretion rate by insulin is associated with hepatic fat content and insulin resistance. Diabetologia. 2007;50(11):2356–65.
Zarei M, et al. Hepatic regulation of VLDL receptor by PPARbeta/delta and FGF21 modulates non-alcoholic fatty liver disease. Mol Metab. 2018;8:117–31.
Adiels M, et al. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2005;25(8):1697–703.
Ekstedt M, et al. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology. 2015;61(5):1547–54.
Geisler CE, Renquist BJ. Hepatic lipid accumulation: cause and consequence of dysregulated glucoregulatory hormones. J Endocrinol. 2017;234(1):R1–21.
Miksztowicz V, et al. Hepatic lipase activity is increased in non-alcoholic fatty liver disease beyond insulin resistance. Diabetes Metab Res Rev. 2012;28(6):535–41.
Taskinen MR, et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler Thromb Vasc Biol. 2011;31(9):2144–50.
Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab. 2020;42:101092.
Fabbrini E, et al. Physiological Mechanisms of Weight Gain-Induced Steatosis in People With Obesity. Gastroenterology. 2016;150(1):79-81 e2.
Acknowledgements
This study thanks to Taiwan Bio-Active Lipid (TBAL) Ltd. Co for assistance in metabolomic analysis.
Funding
CMU109-MF-26, NHRI-EX112-11110BI, MOST 111–2320-B-039–011-, CMU111-MF-91, NSTC 112–2320-B-039–005, MOST 111–2314-B-039–062-MY3, CMU111-MF-41, AUH-11151021, DMR-110–025, MOST 110–2314-B-039–046, DMR-111–204, DMR-112–019, NSTC 111–2622-B-039–004-, DMR-112–189.
Author information
Authors and Affiliations
Contributions
Conceptualization, CR Yang and WL Ma; methodology, CR Yang, PC Shen; validation, PY Liao; formal analysis, CR Yang; investigation, YC Hung, S Mehmood, and WC Chang; resources, HS Lai, WC Cheng, and WL Ma; data curation, WC Chang, HC Lai, S Mehmood, and YC Hung; writing—original draft preparation, CR Yang; writing—review and editing, WL Ma; project administration, WC Cheng; funding acquisition, WC Cheng and WL Ma. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The animal experiments were approved by the Animal Ethics Committee of China Medical University (approval number: CMUIACUC-2021–061). The human study was approved by the Ethics Committee of China Medical University (approval number: CMUH110-REC1-002(CR2)).
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplemental Fig. 1. Trends of total metabolites in mouse models of diet-induced metabolic dysfunction-associated steatohepatitis. (A) Plot generated through principal component analysis (PCA) of the total metabolites of mice fed with NC, HFD, WD, or HFC (16 and 32 wks). PC1: principal component 1; PC2: principal component 2. Each point represents the metabolite profile of a biological replicate. (B) Heatmap exhibiting prominent differences in total metabolites patterns among NC, 16-wk experimental diet, and 32-wk experimental diet. NC: normal chow; HFD: high-fat diet; WD: Western diet; and HFC: high-fat, high-cholesterol diet. Supplemental Table 1. List of real-time PCR primer. Supplemental Table 2. List of all metabolite in human. Supplemental Table 3. List of all lipoprotein subclass of mouse. Supplemental Table 4. List of small metabolites of mouse. Supplemental Table 5. Baseline characteristics of the study cohort.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, CR., Lin, WJ., Shen, PC. et al. Phenotypic and metabolomic characteristics of mouse models of metabolic associated steatohepatitis. Biomark Res 12, 6 (2024). https://doi.org/10.1186/s40364-023-00555-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-023-00555-9