
Abstract
Background
Diabetic foot ulcers (DFU) is the most serious complication of diabetes mellitus, which has become a global health problem due to its high morbidity and disability rates and the poor efficacy of conventional treatments. Thus, it is urgent to identify novel molecular targets to improve the prognosis and reduce disability rate in DFU patients.
Results
In the present study, bulk RNA-seq and scRNA-seq associated with DFU were downloaded from the GEO database. We identified 1393 DFU-related DEGs by differential analysis and WGCNA analysis together, and GO/KEGG analysis showed that these genes were associated with lysosomal and immune/inflammatory responses. Immediately thereafter, we identified CLU, RABGEF1 and ENPEP as DLGs for DFU using three machine learning algorithms (Randomforest, SVM-RFE and LASSO) and validated their diagnostic performance in a validation cohort independent of this study. Subsequently, we constructed a novel artificial neural network model for molecular diagnosis of DFU based on DLGs, and the diagnostic performance in the training and validation cohorts was sound. In single-cell sequencing, the heterogeneous expression of DLGs also provided favorable evidence for them to be potential diagnostic targets. In addition, the results of immune infiltration analysis showed that the abundance of mainstream immune cells, including B/T cells, was down-regulated in DFUs and significantly correlated with the expression of DLGs. Finally, we found latamoxef, parthenolide, meclofenoxate, and lomustine to be promising anti-DFU drugs by targeting DLGs.
Conclusions
CLU, RABGEF1 and ENPEP can be used as novel lysosomal molecular signatures of DFU, and by targeting them, latamoxef, parthenolide, meclofenoxate and lomustine were identified as promising anti-DFU drugs. The present study provides new perspectives for the diagnosis and treatment of DFU and for improving the prognosis of DFU patients.
Similar content being viewed by others

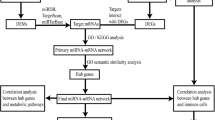

Introduction
DFU is the most common and serious complication in diabetic patients, which is caused by the interaction of multiple risk factors [1]. With the increasing number of diabetic patients worldwide, the incidence of DFUs is also on the rise [2]. The high incidence and disability of DFUs not only lead to a decline in the quality of patients’ lives, but also cause a heavy burden on healthcare and nursing care [3]. It is estimated that between 9.1 and 26.1 million people develop diabetic foot ulcers worldwide each year, with approximately 17% of these cases eventually requiring amputation [4]. It has been shown that a DFU-related amputation occurs every 20 s in the world [5, 6], and the cost of each amputation can be more than $53,500 [7], which imposes a severe financial, physical, and psychological burden on the patient [8]. Currently, the clinical diagnosis of DFU mainly includes the diagnosis of lower limb vasculopathy and peripheral neuropathy [9, 10]. The diagnosis of lower extremity vasculopathy is based on the following criteria: (1) the diagnosis of diabetes mellitus; (2) clinical manifestations of lower extremity ischemia; (3) auxiliary examinations suggesting lower extremity vasculopathy, with an ABI of < 0.9 at rest, or an ABI of > 0.9 at rest, but with discomfort in the lower extremities during exercise, and a decrease in ABI of 15–20% after a plate exercise test, or with stenosis of blood vessels on imaging. The diagnosis of peripheral neuropathy is based on: (1) abnormal temperature sensation; (2) decreased or lost foot sensation in nylon wire examination; (3) abnormal vibration sensation; (4) loss of ankle reflexes; (5) slowing down of 2 or more items of NCV. If two or more of the above five tests are abnormal, the diagnosis is peripheral neuropathy. The clinical diagnosis of DFU mentioned above is complicated and relies heavily on the experience of clinicians, without effective molecular level diagnostic support, that is, there is a lack of effective biomarkers that can be used for clinical diagnosis of DFU. Additionally, there are still limitations in the multiple treatments for DFU, such as treating lower extremity ischemia and foot infections, surgical debridement, intravenous antibiotics, and reducing the pressure of weight-bearing on the ulcers [11,12,13]. Despite recent advances in the treatment of DFUs, a large proportion of patients with DFUs still develop chronic wounds due to irreversible processes [14]. Thus, it is urgent to identify novel biomarkers for its diagnosis and treatment.
Lysosomes are signaling pivots and degradation centers in eukaryotic cells that not only play key roles in processes such as senescence, cellular homeostasis and development; they also act as signaling centers for signal transduction, energy and amino acid sensing, and autophagy regulation [15]. Multiple aspects of these functions that converge on the lysosome are able to connect; when these pathways are unregulated, they become the basis for a wide range of human diseases [16]. It has been shown that relevant lysosomal dysfunction has been found in a variety of diseases including autoimmune, metabolic and renal disorders such as Parkinson’s disease, diabetes mellitus and lysosomal storage disease [17]. Of these, diabetes is one of the leading causes of chronic wound healing problems. When diabetics develop ulcers, they are at high risk for major complications such as infection and amputation [18]. And the DFU is one of the serious complications of diabetes that leads to chronic wound healing problems. Previous studies have shown that lysosomal abnormalities can cause diabetes mellitus, but DFU, as the most common and serious complication of diabetes mellitus, lysosomal abnormalities also have a contributing role in the development of DFU [17]. Thus, in this direction, it is necessary and clinically significant to identify lysosome-related genes that play critical regulatory roles in DFU by whole transcriptome analysis, to explore the molecular changes and functions of lysosome-related genes in the development of DFU, and to ascertain the molecular mechanisms by which lysosome-related genes contribute to the development of DFU.
In the present study, we found that Clusterin (CLU), glutamyl aminopeptidase (ENPEP) and RAB guanine nucleotide exchange factor 1 (RABGEF1) can be indicated by multiple bioinformatic and machine learning algorithms as novel hub lysosome-related genes of DFU (DLGs). Then we constructed a novel artificial neural network (ANN) diagnostic model based on the DLGs to assist in the clinical diagnosis of DFU at the molecular level, and the constructed ANN model showed good diagnostic performance in the training and validation cohorts. Following that, we explored the immune microenvironment in DFU and its relationship with DLGs through immune infiltration analysis performed by the ssGSEA algorithm. Immediately after that, we performed single-cell transcriptome analysis on DFU patients, which revealed that DLGs were also heterogeneously expressed among different cell types, which provided stronger evidence for DLGs as diagnostic target for DFU. Finally, we used molecular docking method to identify drugs that have potential therapeutic or palliative effects on DFU by targeting DLGs. In conclusion, the present study provides new perspectives for understanding the molecular mechanism of DFU and diagnosis and treatment of DFU.
Methods
Data downloading and processing
We downloaded bulk RNA-seq datasets associated with DFU from the GEO database, and datasets that matched the following criteria were included in the present study: Firstly, the dataset is expected to contain unbiased gene expression data, complete annotation information. Second, the DFU patients incorporated into the training cohort and validation cohort should be distinct and independent. Based on the mentioned above criteria, four datasets (GSE80178, GSE134431, GSE7014 and GSE68183) were included in the present study. The basic information of the datasets used in the present study is provided in Table 1. In the present study, GSE80178 and GSE134431 are used as the training cohorts, and GSE7014 and GSE68183 are used as the validation cohorts. Meanwhile, in order to remove the batch effect between datasets caused by different platforms, we used “ComBat” in the R package “sva” to remove the batch effect in the training cohort after merging GSE80178 and GSE134431 and in the validation cohort after merging GSE7014 and GSE68183, respectively [19], and we used principal component analysis (PCA) to assess the effect of de-batching effects. Additionally, the scRNA-seq dataset associated with DFU was obtained from the previous study by Theocharidis, Georgios et al. [20].
Differential analysis of gene expression
The expression profiles of DFUs and Ctrls were compared using the R package “limma”, and dysregulated expression genes (DEGs) were identified in both clusters using a P-value < 0.05 as a criterion.
Weighted correlation network analysis
The “WGCNA” software package was used to identify DFU-related genes in the training cohort [21]. Initially, clustering of samples is performed to detect and exclude outliers to ensure the network analysis is robust. Then constructed the network using a soft thresholding capability to highlight strong correlations and punish weak correlations. Subsequently, we transformed the neighbor-joining matrix into a topological overlap matrix (TOM). Based on the measure of variance of the TOM, and set the minimum module size to 500, and grouped genes with similar expression patterns into the same module by means of average correlation hierarchical grou**. Eventually, we assessed module characterized genes for their correlation with DFUs and identified the matches to the study objectives based on the extent of the correlation.
Protein-protein interaction analysis
We analyzed protein-protein interactions (PPI) between dysregulated lysosomal genes using the STRING database (https://string-db.org/) and Cytoscape software [22]. Dysregulated lysosomal genes were included in the PPI network, and the CytoHubba function in Cytoscape was utilized to provide a composite score for dysregulated lysosomal genes, and the top 40 genes in the composite score were ultimately selected for subsequent analysis.
Functional enrichment analysis
Aiming to ascertain which biological processes and functions the top 40 scoring genes were mainly enriched, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis on the top 40 scoring genes using the R package “clusterProfler” [23]. Pvalue < 0.05 was regarded as statistically significant.
Selection of hub lysosome-related genes of DFU
We used 3 machine algorithms to identify DLGs, namely: least absolute shrinkage and selection operator (LASSO), support vector machine recursive feature elimination (SVM-RFE), and random forest(RF). It has been shown that machine learning has been extensively applied in the biomedical sciences and is capable of efficiently and rapidly analyzing biological data and accurately identifying hub genes in gene expression profiles [Diagnostic evaluation of DLGs for DFU We further investigated whether the selected DLGs had potential value in diagnosing DFU, and thus evaluated the performance of the DLGs. We performed receiver operating characteristic (ROC) analysis using the R software package “pROC” to derive area under roc curve (AUC) values. Specifically, we obtained expression of DLGs and disease state grou** data from DFU sets, performed ROC analysis using the “roc” function of the “pROC” software package, and derived the final AUC results using the “ci” function of “pROC”. We constructed an ANN diagnostic model based on the transcriptome level of DLGs using the R package “neuralnet”. The seed was set to 123 after we normalized the gene expression data using the min-max normalization method. The constructed ANN model consisted of predominantly three layers: 1) Input layer, which mainly consists of the gene expression of the three normalized DLGs. 2) Hidden layer, which mainly includes the gene expressions of the three normalized DLGs and the weights of the three DLGs, as well as the weights between the three hidden layers. 3) Output layer, which indicates the result of judging whether the sample belongs to the Ctrls or the DFUs. We clustered DFUs based on the median expression values of DLGs with gene set enrichment analysis (GSEA) for different subgroups to study which biofunctions and signalling pathways associated with DLGs, and considered P < 0.05 to be statistically significant. Infiltration of immune cells was assessed using ssGSEA [28]. Specifically, ssGSEA was performed in R language using the R packages “GSVA” and “GSEABase” and the immunological characteristics of DFU patients were assessed using the ssGSEA algorithm, respectively. We firstly obtained the information of the genes of 28 immune cells gene sets (Table S1) from the TISIDB database (http://cis.hku.hk/TISIDB/), and then performed single-sample gene set enrichment analysis (ssGSEA) and calculated ssGSEA scores. We used the “pheatmap” R package to visualize the infiltration levels of different immune cells under different infiltration algorithms. To assess the differential infiltration abundance of different immune cells between Ctrls and DFUs, we used the Wilcox test for pairwise comparisons. Subsequently, we used the “ggplot” R package to visualize the infiltration levels of different immune cells under different infiltration algorithms. Subsequently, we used the “ggplot” R package to visualize the correlation between immune cell infiltration abundance and DLGs expression. We downloaded scRNA-seq data of DFU from the previous study by Theocharidis, Georgios et al. [20]. , and analyzed them using the R package “Seurat” [29]. Before analyzing the scRNA-seq data, we weeded out low-quality cells using the following method: cells are likely to be under stress when they have the highest percentage of mitochondrial genes of all genes. Thus, cells with more than 25% of mitochondrial genes will be filtered. Since low-mass cells or empty droplets usually contain less genes and bicellular cells contain more genes, low-mass cells were filtered using the criteria nFeature RNA < 500 and nFeature RNA > 5000. The results yielded 71,718 cells and 14,776 genes. Next, we normalized the gene expression of the cells using the “NormalizeData” function and performed PCA using the ElbowPlot function to extract the top 20 principal components (PCs), which were further analyzed using the “FindVariableFeatures” function. For unsupervised and unbiased clustering of cell subpopulations, we used the “FindNeighbors”, “FindClusters” (resolution = 0.5) and “RunUMAP” functions. We then annotated the cell types using known markers [30,Full size imageConstruction and verification of the artificial neural network model
Single-gene GSEA analysis
Evaluation of immune cell infiltration and their correlation with the DLGs
scRNA-seq analysis
Identification and docking potential drugs that target DLGs
The previous studies have shown that the process of drug discovery begins with the identification of disease targets, and target-based drug discovery is the most common strategy for new drug development [42, 43]. The mentioned above showed that DLGs are promising as diagnostic targets for DFU, which means that we can also identify drugs that can treat or alleviate DFU by targeting DLGs. Specifically, we used the Enrichr platform (https://maayanlab.cloud/Enrichr/) for online analysis and identification. According to the DSigDB database, we identified four drugs that were able to target DLGs with p-values less than 0.05. Immediately following, we investigated the binding affinity of the selected drugs to DLGs using molecular docking techniques. The results showed that all four drugs were able to target CLU and the binding energies were relatively high, respectively, CLU-latamoxef (-7.1 kcal/mol), CLU-parthenolide (-6.0 kcal/mol), CLU-meclofenoxate (-5.1 kcal/mol) and CLU- lomustine (-4.5 kcal/mol) (Fig. 8A-D). Subsequently, latamoxef and meclofenoxate were both able to target ENPEP, respectively ENPEP-latamoxef (-8.6 kcal/mol) and ENPEP-meclofenoxate (-5.9 kcal/mol) (Fig. 8E-F). Additionally, parthenolide and lomustine were also able to target RABGEF1, respectively RABGEF1-parthenolide (-6.3 kcal/mol) and RABGEF1-lomustine (-5.1 kcal/mol) (Fig. 8G-H). Taken together, the docking results suggest that these potentially targeted drugs may influence the development of DFU by interacting with DLGs to treat or alleviate the DFU symptoms.

The 3D structures of complexes formed by the interaction of selected drugs with DLGs. (A-D)The structure of the complex formed by the docking of CLU with latamoxef (A), parthenolide (B), meclofenoxate (C) and lomustine (D). (E-F) The structure of the complex formed by the docking of ENPEP with latamoxef (E) and meclofenoxate (F). (G-H). The structure of the complex formed by the docking of RABGEF1 with parthenolide (G) and lomustine (H)
