
Abstract
Background
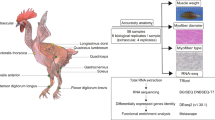
Broilers stand out as one of the fastest-growing livestock globally, making a substantial contribution to animal meat production. However, the molecular and epigenetic mechanisms underlying the rapid growth and development of broiler chickens are still unclear. This study aims to explore muscle development patterns and regulatory networks during the postnatal rapid growth phase of fast-growing broilers. We measured the growth performance of Cornish (CC) and White Plymouth Rock (RR) over a 42-d period. Pectoral muscle samples from both CC and RR were randomly collected at day 21 after hatching (D21) and D42 for RNA-seq and ATAC-seq library construction.
Results
The consistent increase in body weight and pectoral muscle weight across both breeds was observed as they matured, with CC outpacing RR in terms of weight at each stage of development. Differential expression analysis identified 398 and 1,129 genes in the two dimensions of breeds and ages, respectively. A total of 75,149 ATAC-seq peaks were annotated in promoter, exon, intron and intergenic regions, with a higher number of peaks in the promoter and intronic regions. The age-biased genes and breed-biased genes of RNA-seq were combined with the ATAC-seq data for subsequent analysis. The results spotlighted the upregulation of ACTC1 and FDPS at D21, which were primarily associated with muscle structure development by gene cluster enrichment. Additionally, a noteworthy upregulation of MUSTN1, FOS and TGFB3 was spotted in broiler chickens at D42, which were involved in cell differentiation and muscle regeneration after injury, suggesting a regulatory role of muscle growth and repair.
Conclusions
This work provided a regulatory network of postnatal broiler chickens and revealed ACTC1 and MUSTN1 as the key responsible for muscle development and regeneration. Our findings highlight that rapid growth in broiler chickens triggers ongoing muscle damage and subsequent regeneration. These findings provide a foundation for future research to investigate the functional aspects of muscle development.
Similar content being viewed by others

Introduction
Chicken, a pivotal source of animal high-quality protein in human diets, stands as one of the most widely consumed food products globally [1]. Over the past few decades, a substantial surge in body weight and pectoral muscle weight has been witnessed, mainly attributable to intensive genetic selection [2, 3]. Skeletal muscle, accounting for about 40% of body mass of animals, is comprised of elongated, multinucleated muscle fibers [4, 5]. Multinucleated muscle fibers are formed from the fusion of myoblasts during embryonic myogenesis [6]. The number of myofibers in animals typically remains unchanged after birth, with muscle growth primarily depending on myofiber hypertrophy [7]. The growth and development of skeletal muscle is a complex biological process that is regulated by diverse genes, transcription factors, and epigenetic factors [8]. Revealing the molecular mechanisms of animal muscle development has been a focus of research in the field of genetic and breeding research.
With the advancements in high-throughput sequencing and multi-omics technology, unraveling the intrinsic biological mechanisms of muscle development has become more convenient and cost-effective [9, 10]. An increasing number of genes or quantitative trait loci that regulate the growth traits of chickens have been characterized. A recent analysis from genome-wide association studies (GWAS) identified ADGRG6 as a major candidate gene responsible for carcass composition traits [11]. The genes NSUN3 and AGK were related to muscle development in broiler chickens. Despite the decreasing sequencing costs, GWAS remain costly, necessitating large sample sizes to gain sufficient SNP data [12, 13]. Challenges like linkage disequilibrium and population stratification complicate the accurate identification of causal variants in GWAS [14]. Further research at the transcriptome level could provide more comprehensive mechanistic insights [15, 16]. Comparison studies between modern broiler chickens and local breeds, conducted through RNA sequencing have revealed the upregulated genes such as IGF1, IGF1R, WFIKKN2, positively regulating chicken growth [17]. In another study using fast- and slow-growth meat chickens, DNA methylation analysis identified methylation sites in growth factors like IGF1R and FGF18, which are related to skeletal muscle development [18]. In 2013, the assay for transposase accessible chromatin with high-throughput sequencing (ATAC-seq) was developed for evaluating genome-wide chromatin accessibility and identifying transcription factor binding sites [19]. Integrating ATAC-seq with high-throughput paired-end RNA sequencing (RNA-seq) data can offer more precise information on the impact of possible regulatory differences in differentially enriched open chromatin regions [1: Table S2). The mapped reads were counted at gene level using featureCount (SUBREAD package; v.1.6.3) [27] with the chicken reference (GRCg7b) and normalized using TPM procedure to compute the TPM values for subsequent analysis steps. Principal component analysis (PCA) was performed using R package FactoMineR (v.2.9.0) [28]. Differentially expressed genes (DEGs) between different groups were identified by DESeq2 package (v.1.36.0) [29] in the R program. Genes with an adjusted P value < 0.05 and a log2 (Fold Change) > |1| were considered significant DEGs [30,1: Table S3). Subsequently, duplicate reads were marked and removed using Sambamba (v.0.7.1) [36]. The insert length was directly calculated from the aligned BAM file with Samtools (v.0.1.11) [37]. For peak calling, MACS2 (v.2.2.7.1) [38] was employed. IDR (v.2.0.3) was used to filter enriched regions at an IDR of less than 0.05 to generate high-confidence peaks [39]. Peak annotation and analysis were carried out using ChIPseeker (v.1.32.1) [40]. Promoter regions were defined as peaks overlap** a region that was ± 3 kb from the transcriptional start site (TSS). Differential peak analysis was performed using the diffBind package [41] with the false discovery ratio < 0.05 and log2 (Fold Change) > |0.5| in two comparison groups [42,43,44]. The Bedtools (v.2.30.0) [45] suite was employed to calculate overlap and enrichment between different intervals. The peaks of different groups were visualized in IGV tool (v.2.13.2) [46, 47].
Integrative analysis of RNA-seq and ATAC-seq data
Since chromatin accessibility is closely related to gene regulation, the VennDiagram package (v.1.7.3) [48] in R was used to obtain the overlap** genes of DEGs derived from RNA-seq and genes associated with differential accessible regions (DARs) identified in ATAC-seq. Subsequently, the clusterProfiler [33] was utilized for GO enrichment function analysis and KEGG pathway analysis of these overlap** genes. The genes of top 10 terms were extracted for frequency statistics. We combined the genes with higher frequencies into gene sets, which were used as input for signal pathway and network analysis on the GeneMANIA [49] and STRING online platforms [50].
Results
Time-course growth performance analysis of broilers
We monitored the growth performance of CC and RR over a 42-d period, including the BW and PMW, and examined their morphology of muscle fiber from D7 to D42. As expected, the results showed that BW and PMW of the two groups gradually increased with advancing age while the weight of CC was significantly higher than RR at all growth stages (Fig. 1a and b, Additional file 1: Table S4). The BW and PMW of CC group were 2,986.70 ± 25.61 g and 493.76 ± 76.13 g at D42. The BW and PMW of RR were 2,460.93 ± 168.09 g and 369.03 ± 52.95 g at the same time. The histological examination revealed minor differences between CC and RR groups with CSA and MFD (P > 0.05) except the CSA at D21 and D35 (Fig. 1c and d) while the slice diagram revealed that the size of muscle fibers continued to increase with age. Morphology of the myofibers at all time point after hatching is shown here (Fig. 1e). The BW and PMW were positively correlated with CSA while negatively correlated with MFD of myofibers at D21 and D42 (Fig. 1f). When considering the weight results, it became evident that both BW and PMW showed a gradual increase before D21, followed by a linear increase after D21. These results suggested that D21 and D42 are crucial stages for broiler growth. To identify the genes that were differentially expressed between different groups, pectoral muscle tissue of CC and RR was collected at D21 and D42 for RNA-seq and ATAC-seq (Fig. 1g).

Growth performance of postnatal broiler chickens. a The changes in body weight (BW) of individuals with Cornish (CC) and White Plymouth Rock (RR) from the day of hatching (DOH) and day 7 after hatching (D7), D14, D21, D28, D35, D42. b The changes in pectoral muscle weight (PMW) of individuals with CC and RR from DOH to D42. c Cross-sectional area (CSA) of muscle fibers from D7 to D42. d Muscle fiber density (MFD) changes from D7 to D42. e Morphology of the myofibers stained by hematoxylin–eosin from D7 to D42. The corresponding scale is marked in the lower left corner of each image. Scale bars: 100 μm. f The scatter plot and Pearson’s correlation of broiler chickens at D21 and D42. g Pectoral muscles were collected at D21 and D42 for RNA-seq and ATAC-seq analysis. ** and * indicate P value less than 0.01 and 0.05, respectively
Transcriptional profiling of pectoral muscle
The PCA plot exhibited a separation between the two stages of D21 and D42 along PC2 (Fig. 2a). To identify the genes that were DEGs between different groups, we performed a differential expression analysis with the four groups in the two dimensions of breeds and ages. The volcano map preliminarily showed the expression of DEGs, as shown in Fig. 2b. A total of 204, 219 D21BGs and 302, 404 D42BGs of CC and RR were identified, respectively (Additional file 1: Table S4–5). Additionally, there were 137, 58 CBGs and 143, 60 RBGs at D21 and D42 (Additional file 1: Table S5–6).

Analysis of RNA-seq data from the pectoral muscle. a PCA plot of RNA-seq data of CC and RR groups at D21 and D42. Each point represents a sample. b Volcano plot of differential expressed genes (DEGs) obtained through the comparison between pairwise groups. c The GO enrichment results of CC-biased DEGs (CBGs) and RR-biased DEGs (RBGs) at D21 and D42. d GO enrichment results of D21-biased DEGs (D21BGs) and D42-biased DEGs (D42BGs) of CC and RR
In terms of differential gene functions between CC and RR groups at D21 and D42, various biological functions were enriched. At D21, CBGs were primarily enriched in terms of extracellular space and anchored component of membrane (Fig. 2c). Additionally, they were also enriched in Glycolysis/Gluconeogenesis, Fructose and mannose metabolism, Purine metabolism, and Cell adhesion molecules (Additional file 2: Fig. S1a). Among them, ALDOB were found to be related to both the extracellular space term and the Glycolysis/Gluconeogenesis pathway. RBGs at D21 were enriched in the terms such as circadian rhythm and negative regulation of cellular response to growth factor stimulus (Fig. 2c). The CBGs at D42 were enriched in ATP-dependent chromatin remodeling and WNT signaling pathway as shown in Fig. S1a. Furthermore, the pathway of Purine metabolism, including NPR1, was identified at both CBGs at D21 and D42. The RBGs at D42 were primarily enriched in cholesterol metabolic process term and amino acid metabolism pathways (Fig. 2c, Additional file 2: Fig. S1a).
A total of 137, 58 CBGs and 143, 60 RBGs at D21 and D42 were identified, respectively (Additional file 1: Table S7–8). Significant enrichment was observed in terms associated with apical plasma membrane and actin-based cell projection of D21BGs of CC (Fig. 2d). The D42BGs of CC were mainly involved in cell differentiation and muscle development, including FOS, MUSTN1, TGFB3, WNT9A, and GREM1 (Fig. 2d). Additionally, the D21BGs of RR were enriched in terms such as myosin complex and cytoskeletal motor activity that included MYH1D, MYO1A, and ACTC1. The D42BGs in RR exhibited enrichment in defense response and immune system processes, as well as pathways such as cytokine-cytokine receptor interaction and cell adhesion molecules (Fig. 2d, Additional file 2: Fig. S1b). Notably, D21BGs and D42BGs of CC and RR were involved in steroid biosynthesis and cell–cell interactions (Additional file 2: Fig. S1b).
Analysis of chromatin accessibility in pectoral muscle
In order to better understand the mechanisms underlying gene expression alterations, we conducted a comprehensive examination of chromatin openness using ATAC-seq across the four experimental groups. The ATAC-seq data displayed approximately 200 bp fragment insert size (Fig. 3a, Additional file 2: Fig. S2a–c), and the PCA revealed a separation tendency among the four groups (Fig. 3b), indicating the reliability of the ATAC-seq data. For the 12 pectoral muscle samples, a total of 75,149 ATAC-seq peaks were identified (Additional file 2: Fig. S2d). These peaks were annotated in their promoter, exon, intron and intergenic regions, and a higher abundance observed in promoter and intronic regions (Fig. 3c). Contrasting the peaks in terms of breed and time dimensions revealed a comparable number of intersecting peaks between the two groups. Specifically, the intersection of peaks between CC and RR at D21 and D42 were 11,418 and 11,151, respectively (Fig. 3d). Furthermore, the intersection of peaks between pectoral muscle samples of D21 and D42 in the CC and RR groups were 11,681 and 10,843, respectively (Fig. 3d).

Analysis of chromatin accessibility from the pectoral muscle. a Fragment insert length distribution plots of ATAC-seq samples. b PCA plot of ATAC-seq data of CC and RR groups at D21 and D42. Each point represents a sample. c The feature distribution of ATAC-seq peak dataset. d Overlap peaks between two groups. e Heatmaps of differential accessible regions (DARs)
Subsequently, a pairwise comparative analysis based on the ATAC-seq data was conducted to identify DARs. A total of 3,283, 24,417, 7,916, and 15,553 significant DARs were identified in D21, D42, CC and RR development (Additional file 1: Table S9–12). The genes nearest DARs were annotated, revealing 1,486, 8,462, 3,028, and 5,615 genes at D21, D42, CC, and RR, respectively. The heatmaps revealed that these significant DARs showed very clear specific patterns in each group (Fig. 3e, Additional file 2: Fig. S2e).
Integration analysis of ATAC-seq and RNA-seq
To further determine the relationship between gene expression and open chromatin regions, we examined the association between gene accessibility and gene expression during pectoral muscle development by integrating RNA-seq and ATAC-seq data (Additional file 1: Table S13). As the Venn diagrams shown, we found 6 upregulated CBGs carrying DARs, including TMEM164, FSTL4, ZNF692, KIF14, APOA5, and KIFC1, and 16 RBGs carrying DARs related to muscle development, such as MYO7B at D21 (Fig. 4a). In addition, 12 upregulated and 1 downregulated DEGs contained DARs were identified in the CC vs. RR at D42. Among these, the RBG was identified as IRF9, which was associated with the inflammatory response. Focusing on the impact of age on chromatin accessibility, we identified 69 D42BGs carrying DARs of CC group, including WNT9A, TGFB3 that related to muscle development. Additionally, we found 25 D21BGs containing DARs in CC groups, which included NRG1, a gene related to lipid metabolism, and TMEM164, a member of the transmembrane protein family. Moreover, 5 upregulated DEGs carrying DARs were found between D21 and D42 of RR, including IRF7, DCSTAMP, RGCC, TTC7A and NR1D1. In this comparison, there were 65 DEGs with DARs were downregulated at D42. As expected, a positive correlation was found between accessibility signatures and gene expression patterns, based on the calculated fold changes by assigning open chromatin regions to the nearest DEGs (Fig. 4b, Additional file 2: Fig. S2f).

Integration of RNA-seq and ATAC-seq to identify candidate genes. a Venn diagrams showed the DEGs carrying DARs. b The relationship between DEGs and DARs. c The 16 RBGs at D21 and the 65 D21BGs of RR were combined for GO enrichment analysis. d Frequency analysis of DEGs of top 20 terms. e The network analysis and functional enrichment analysis of the DEGs was performed using GeneMANIA. f Differential expression results among the four groups. ** and * indicate P value less than 0.01 and 0.05, respectively
Insights into muscle development at D21 in broilers
Based on the data showed in Fig. 4a, it was determined that there were four distinct classes of DEGs exhibiting higher count level, including CC-biased genes at D42, RR-biased genes at D21, D42-biased genes of CC, and D21-biased genes of RR, within the eight identified clusters. Considering the differential expression patterns, we have merged a set of 16 RR-biased genes at D21 with 65 D21-biased genes of CC. This combination of DEGs, resulting in a total of 81 DEGs that were highly expressed in RR at D21, for subsequent analysis. Enrichment analysis indicated that these genes were mainly involved in biological processes such as cholesterol metabolic process and actin filament-based movement (Fig. 4c). For further analysis, the DEGs of top 20 terms were chosen for frequency analysis, and the 14 genes were ranked (Fig. 4d). It was observed that FDPS and CHRM4 had the highest frequency, appearing 6 times, followed by MTNR1A, CETP, PRPS2, VTG1, HBBA and HBA1, indicating their crucial role in the development of RR at D21. The subsequent analysis was conducted using the GeneMANIA, which confirmed the strong association of ACTC1, NRG1, and MYC with mesenchyme development, and FDPS and CETP with sterol metabolic process (Fig. 4e).
The gene expression profiles in broiler chickens at both D21 and D42 are depicted in Fig. 4f. The findings indicated a significant upregulation of ACTC1, HBBA, HBA1, FDPS, and NRG1 in broiler chickens at D21 compared to D42. It is known that the rapid muscle development occurs in broiler chickens during the D21 period. Based on these observations, it can be hypothesized that ACTC1 and NRG1 play crucial roles in muscle development during this period, while FDPS, HBBA and HBA1 may exert a coordinated regulatory influence on muscular development.
Muscle regulation dynamics and regeneration mechanisms at D42
On the flip side, we noted that the 12 CBGs at D42 and 69 D42BGs of CC could be classified into a singular category that were highly expressed in CC at D42. This classification facilitated an in-depth exploration of the regulatory patterns governing muscle development in broiler chickens at D42, resulting in the identification of 81 DEGs. Subsequent functional enrichment analysis of these 81 DEGs revealed their significant involvement in organ developmental process, cell differentiation, and SMAD protein signal transduction (Fig. 5a). Using the method described above, we selected the top 10 terms for frequency analysis of the DEGs, and the resulting 9 genes were ranked accordingly (Fig. 5b). Notably, TGFB3 exhibited the highest frequency, appearing 8 times, followed by FOS, WNT9A, HDAC9, and MUSTN1, underscoring their pivotal role in the development of CC at D42.

Intense muscle damage with rapid muscle growth at D42. a The 12 CBGs at D42 and the 69 D42BGs of CC were combined for GO enrichment analysis. b Frequency analysis of 9 DEGs of top 10 terms. c Network analysis and functional enrichment analysis using GeneMANIA on the 9 DEGs that were present in at least 2 terms among the 20 DEGs. d The network analysis of DEGs was performed using STRING. e Differential expression results among the four groups. ** and * indicate P value less than 0.01 and 0.05, respectively. f Gene expression and chromatin accessibility of FOS and MUSTN1 displayed similar dynamic changes in CC at D42
Utilizing GeneMANIA, we conducted the network analysis and functional enrichment analysis on the 9 DEGs in Fig. 5c. The results revealed that LMOD2 was associated with myofiber contraction and cytoskeletal organization. Protein–protein interaction analysis revealed that LMOD2 interacts with MUSTN1 and MB (Fig. 5d). This finding suggested a potential functional relationship between these DEGs, which could be significant in the context of muscle development and function. Notably, we observed that MUSTN1, FOS, TGFB3, and WNT9A were highly expressed in CC at D42 among the four groups (Fig. 5e). Furthermore, the gene expression and chromatin accessibility of FOS and MUSTN1, which are related to muscle regeneration, exhibited similar dynamic changes in CC at D42 (Fig. 5f).
Discussion
As a crucial tissue in meat-producing animals, skeletal muscle plays a vital role in initiating movements, supporting respiration, and maintaining homeostasis [51, 52]. Chickens serve as significant scientific models for studying skeletal muscle development in animals, given that the progression of chicken anatomy closely mirrors that of mammals [53, 54]. Through prolonged selective breeding practices, broiler chickens have developed an exceptionally rapid growth rate, primarily attributed to the prioritization of the pectoral muscle’s weight as a breeding goal, which rapidly increases in size after hatching [55, 86, 87]. It is noteworthy that broiler chickens exhibit sustained accelerated muscle growth at D42, which may be closely related to the elevated expression of TGFB3. Importantly, the study revealed the presence of several genes associated with muscle regeneration, such as FOS and MUSTN1, in broilers at D42. Research has shown that FOS plays a crucial role in initiating key stem cell functions including cell cycle entry, proliferative expansion, and muscle regeneration [88]. The function of MUSTN1 in skeletal muscle physiology is multifaceted, with the knockdown of MUSTN1 has been shown to induce changes in myofiber types [89] and to exert influences on both myoblast differentiation and muscle regeneration processes [90]. Myoblast differentiation is impaired and the fusion of myoblasts fails to occur with the absence of MUSTN1. In-depth vivo experiments have demonstrated that MUSTN1 functions as an early marker of myoblast fusion, playing a role in skeletal muscle development and regeneration [90]. Furthermore, inhibition of MUSTN1 expression impairs myoblast differentiation by targeting MYOD and MYOG [91]. Interestingly, our H&E staining results of muscle results showed that numerous myonuclei within the muscle fibers of broilers during the growth period were located centrally rather than at the periphery, suggesting a state of continuous muscle repair [92, 93]. This further indicates that intense muscle damage may coincide with rapid muscle growth until D42. To ensure continuous muscle mass growth, the organism responds by upregulating FOS and MUSTN1, facilitating repair and achieving a stable state of muscle differentiation in the body.
Conclusion
This research provides a high-resolution exploration of the transcriptome and chromatin accessibility landscapes of pectoral muscle in broiler chickens. Our study explores the developmental dynamics of the pectoral muscle, focusing on various dimensions and the results underscore that temporal differences outweigh those observed in the breed dimension. Moreover, our findings illuminate that the rapid growth observed in broiler chickens induces continuous muscle damage followed by subsequent regeneration. Key candidate genes, including ACTC1, FDPS, NRG1, TGFB3, MUSTN1 and FOS, were identified to play crucial roles in the rapid post-hatching development of broiler chickens. Overall, our research contributes to a deeper understanding of the regulatory mechanisms governing late-stage muscle development in broiler chickens. Additionally, the identification of key candidate genes lays a foundation for potential future studies delving into the functional aspects of muscle development.
Availability of data and materials
The datasets generated and analyzed during the current study are not publicly available. They can be made available from the corresponding author upon reasonable request.
Abbreviations
- ACTC1:
-
Actin alpha cardiac muscle 1
- ATAC-seq:
-
Assay for transposase accessible chromatin with high-throughput sequencing
- BW:
-
Body weight
- CBGs:
-
CC-biased DEGs
- CC:
-
Cornish
- CETP:
-
Cholesteryl ester transfer protein
- CSA:
-
Cross-sectional area
- DOH:
-
Day of hatching
- D7:
-
Day 7 after hatch
- DARs:
-
Differential accessible regions
- DEGs:
-
Differentially expressed genes
- D21BGs:
-
D21-biased DEGs
- D42BGs:
-
D42-biased DEGs
- FDPS:
-
Farnesyl diphosphate synthase
- FOS:
-
Fos proto-oncogene, AP-1 transcription factor subunit
- GO:
-
Gene ontology
- GWAS:
-
Genome-wide association studies
- HBA1:
-
Hemoglobin subunit alpha 1
- HBBA:
-
Hemoglobin subunit epsilon 1
- H&E:
-
Hematoxylin–eosin
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- MFD:
-
Muscle fiber density
- MUSTN1:
-
Musculoskeletal, embryonic nuclear protein 1
- NRG1:
-
Neuregulin 1
- PCA:
-
Principal component analysis
- PMW:
-
Pectoral muscle weight
- RBGs:
-
RR-biased DEGs
- RNA-seq:
-
High-throughput paired-end RNA sequencing
- RR:
-
White Plymouth Rock
- TGFB3:
-
Transforming growth factor beta 3
- TMEM164:
-
Transmembrane protein 164
- TSS:
-
Transcriptional start site
- WNT9A:
-
Wnt family member 9A
References
Huo W, Weng K, Li Y, Zhang Y, Zhang Y, Xu Q, et al. Comparison of muscle fiber characteristics and glycolytic potential between slow- and fast-growing broilers. Poult Sci. 2022;101(3):101649. https://doi.org/10.1016/j.psj.2021.101649.
Grodzik M, Sawosz F, Sawosz E, Hotowy A, Wierzbicki M, Kutwin M, et al. Nano-nutrition of chicken embryos. The effect of in ovo administration of diamond nanoparticles and l-glutamine on molecular responses in chicken embryo pectoral muscles. Int J Mol Sci. 2013;14(11):23033–44. https://doi.org/10.3390/ijms141123033.
Ahmadipour B, Khajali F. Expression of antioxidant genes in broiler chickens fed nettle (Urtica dioica) and its link with pulmonary hypertension. Anim Nutr. 2019;5(3):264–9. https://doi.org/10.1016/j.aninu.2019.04.004.
Yin H, Price F, Rudnicki MA. Satellite cells and the muscle stem cell niche. Physiol Rev. 2013;93(1):23–67. https://doi.org/10.1152/physrev.00043.2011.
Lavin KM, Roberts BM, Fry CS, Moro T, Rasmussen BB, Bamman MM. The importance of resistance exercise training to combat neuromuscular aging. Physiology (Bethesda). 2019;34(2):112–22. https://doi.org/10.1152/physiol.00044.2018.
Mishra P, Varuzhanyan G, Pham AH, Chan DC. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab. 2015;22(6):1033–44. https://doi.org/10.1016/j.cmet.2015.09.027.
Gu JM, Wang DJ, Peterson JM, Shintaku J, Liyanarachchi S, Coppola V, et al. An NF-κB–Ephrina5-dependent communication between NG2+ interstitial cells and myoblasts promotes muscle growth in neonates. Dev Cell. 2016;36(2):215–24. https://doi.org/10.1016/j.devcel.2015.12.018.
Chen M, Wei X, Song M, Jiang R, Huang K, Deng Y, et al. Circular RNA circMYBPC1 promotes skeletal muscle differentiation by targeting MyHC. Mol Ther Nucleic Acids. 2021;24:352–68. https://doi.org/10.1016/j.omtn.2021.03.004.
Zhang Y, Li Y, Chen X, Meng Z, Guo S. Combined metabolome and transcriptome analyses reveal the effects of mycorrhizal fungus Ceratobasidium sp. AR2 on the flavonoid accumulation in Anoectochilus roxburghii during different growth stages. Int J Mol Sci. 2020;21(2):564. https://doi.org/10.3390/ijms21020564.
Wang X, Wen Y. A penalized linear mixed model with generalized method of moments for prediction analysis on high-dimensional multi-omics data. Brief Bioinform. 2022;23(4):bbac193. https://doi.org/10.1093/bib/bbac193.
Yang X, Sun J, Zhao G, Li W, Tan X, Zheng M, et al. Identification of major loci and candidate genes for meat production-related traits in broilers. Front Genet. 2021;12:645107. https://doi.org/10.3389/fgene.2021.645107.
Moon S, Kim YJ, Han S, Hwang MY, Shin DM, Park MY, et al. The korea biobank array: design and identification of coding variants associated with blood biochemical traits. Sci Rep. 2019;9:1382. https://doi.org/10.1038/s41598-018-37832-9.
Xu J, Zhao Z, Zhang X, Zheng X, Li J, Jiang Y, et al. Development and evaluation of the first high-throughput SNP array for common carp (Cyprinus carpio). BMC Genomics. 2014;15:307. https://doi.org/10.1186/1471-2164-15-307.
Visscher PM, Brown MA, Mccarthy MI, Yang J. Five years of gwas discovery. Am J Hum Genet. 2012;90(1):7–24. https://doi.org/10.1016/j.ajhg.2011.11.029.
Li W, Zheng M, Zhao G, Wang J, Liu J, Wang S, et al. Identification of qtl regions and candidate genes for growth and feed efficiency in broilers. Genet Sel Evol. 2021;53:13. https://doi.org/10.1186/s12711-021-00608-3.
Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Map** and quantifying mammalian transcriptomes by RNA-seq. Nat Methods. 2008;5(7):621–8. https://doi.org/10.1038/nmeth.1226.
Davis RV, Lamont SJ, Rothschild MF, Persia ME, Ashwell CM, Schmidt CJ. Transcriptome analysis of post-hatch breast muscle in legacy and modern broiler chickens reveals enrichment of several regulators of myogenic growth. PLoS One. 2015;10(3):e122525. https://doi.org/10.1371/journal.pone.0122525.
Hu Y, Xu H, Li Z, Zheng X, Jia X, Nie Q, et al. Comparison of the genome-wide dna methylation profiles between fast-growing and slow-growing broilers. PLoS One. 2013;8(2):e56411. https://doi.org/10.1371/journal.pone.0056411.
Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods. 2013;10(12):1213–8. https://doi.org/10.1038/nmeth.2688.
Yue D, Liu W, Gao L, Zhang L, Wang T, **ao S, et al. Integrated multiomics analyses revealing different molecular profiles between early- and late-stage lung adenocarcinoma. Front Oncol. 2021;11:746943. https://doi.org/10.3389/fonc.2021.746943.
Shen X, Liu Z, Cao X, He H, Han S, Chen Y, et al. Circular RNA profiling identified an abundant circular RNA circTMTC1 that inhibits chicken skeletal muscle satellite cell differentiation by sponging miR-128-3p. Int J Biol Sci. 2019;15(10):2265–81. https://doi.org/10.7150/ijbs.36412.
Mai C, Wen C, Xu Z, Xu G, Chen S, Zheng J, et al. Genetic basis of negative heterosis for growth traits in chickens revealed by genome-wide gene expression pattern analysis. J Anim Sci Biotechnol. 2021;12:52. https://doi.org/10.1186/s40104-021-00574-2.
Van Every HA, Schmidt CJ. Transcriptomic and metabolomic characterization of post-hatch metabolic reprogramming during hepatic development in the chicken. BMC Genomics. 2021;221:380. https://doi.org/10.1186/s12864-021-07724-w.
Gu S, Wen C, **ao Z, Huang Q, Jiang Z, Liu H, et al. Myov: a deep learning-based tool for the automated quantification of muscle fibers. Brief Bioinform. 2024;25(2):bbad528. https://doi.org/10.1093/bib/bbad528.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one fastq preprocessor. Bioinformatics. 2018;34(17):i884–90. https://doi.org/10.1093/bioinformatics/bty560.
Kim D, Langmead B, Salzberg SL. Hisat: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–60. https://doi.org/10.1038/nmeth.3317.
Liao Y, Smyth GK, Shi W. Featurecounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–30. https://doi.org/10.1093/bioinformatics/btt656.
Lê S, Josse J, Husson F. Factominer: an R package for multivariate analysis. J Stat Softw. 2008;25(1):1–18. https://doi.org/10.18637/jss.v025.i01.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. https://doi.org/10.1186/s13059-014-0550-8.
Dunn SJ, Li MA, Carbognin E, Smith A, Martello G. A common molecular logic determines embryonic stem cell self-renewal and reprogramming. EMBO J. 2019;38(1):e100003. https://doi.org/10.15252/embj.2018100003.
Marinov GK, Trevino AE, **ang T, Kundaje A, Grossman AR, Greenleaf WJ. Transcription-dependent domain-scale three-dimensional genome organization in the dinoflagellate breviolum minutum. Nat Genet. 2021;53(5):613–7. https://doi.org/10.1038/s41588-021-00848-5.
Redmon IC, Ardizzone M, Hekimoğlu H, Hatfield BM, Waldern JM, Dey A, et al. Sequence and tissue targeting specificity of ZFP36L2 reveals Elavl2 as a novel target with co-regulation potential. Nucleic Acids Res. 2022;50(7):4068–82. https://doi.org/10.1093/nar/gkac209.
Yu G, Wang LG, Han Y, He QY. Clusterprofiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. https://doi.org/10.1089/omi.2011.0118.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics. 2014;30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short dna sequences to the human genome. Genome Biol. 2009;10(3):R25. https://doi.org/10.1186/gb-2009-10-3-r25.
Tarasov A, Vilella AJ, Cuppen E, Nijman IJ, Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31(12):2032–4. https://doi.org/10.1093/bioinformatics/btv098.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. https://doi.org/10.1093/bioinformatics/btp352.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. https://doi.org/10.1186/gb-2008-9-9-r137.
Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, et al. Chip-seq guidelines and practices of the encode and modencode consortia. Genome Res. 2012;22(9):1813–31. https://doi.org/10.1101/gr.136184.111.
Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31(14):2382–3. https://doi.org/10.1093/bioinformatics/btv145.
Stark R, Brown G. Diffbind: differential binding analysis of chip-seq peak data. 2012.
Hatzi K, Geng H, Doane AS, Meydan C, Lariviere R, Cardenas M, et al. Histone demethylase LSD1 is required for germinal center formation and BCL6-driven lymphomagenesis. Nat Immunol. 2019;20:86–96. https://doi.org/10.1038/s41590-018-0273-1.
Fang R, Preissl S, Li Y, Hou X, Lucero J, Wang X, et al. Comprehensive analysis of single cell atac-seq data with snapatac. Nat Commun. 2021;12:1337. https://doi.org/10.1038/s41467-021-21583-9.
Kinoshita M, Barber M, Mansfield W, Cui Y, Spindlow D, Stirparo GG, et al. Capture of mouse and human stem cells with features of formative pluripotency. Cell Stem Cell. 2021;28(3):453–71. https://doi.org/10.1016/j.stem.2020.11.005.
Quinlan AR, Hall IM. Bedtools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2. https://doi.org/10.1093/bioinformatics/btq033.
Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48(3):273–82. https://doi.org/10.1038/ng.3500.
Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m6A-guided epigenetic inhibition of LYPD1. Mol Cancer. 2020;19:123. https://doi.org/10.1186/s12943-020-01239-w.
Chen H, Boutros PC. Venndiagram: a package for the generation of highly-customizable venn and euler diagrams in R. BMC Bioinformatics. 2011;12:35. https://doi.org/10.1186/1471-2105-12-35.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(suppl_2):W214–20. https://doi.org/10.1093/nar/gkq537.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. String v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52. https://doi.org/10.1093/nar/gku1003.
Schroder EA, Esser KA. Circadian rhythms, skeletal muscle molecular clocks, and exercise. Exerc Sport Sci Rev. 2013;41(4):224–9. https://doi.org/10.1097/JES.0b013e3182a58a70.
Hernandez CA, Gonzales NM, Parker CC, Sokolof G, Vandenbergh DJ, Cheng R, et al. Genome-wide associations reveal human-mouse genetic convergence and modifiers of myogenesis, CPNE1 and STC2. Am J Hum Genet. 2019;105(6):1222–36. https://doi.org/10.1016/j.ajhg.2019.10.014.
Zhang D, Ran J, Li J, Yu C, Cui Z, Amevor FK, et al. miR-21–5p regulates the proliferation and differentiation of skeletal muscle satellite cells by targeting KLF3 in chicken. Genes (Basel). 2021;12(6):814. https://doi.org/10.3390/genes12060814.
Han S, Cui C, Zhao X, Zhang Y, Zhang Y, Zhao J, et al. Filamin C regulates skeletal muscle atrophy by stabilizing dishevelled-2 to inhibit autophagy and mitophagy. Mol Ther Nucleic Acids. 2022;27:147–64. https://doi.org/10.1016/j.omtn.2021.11.022.
Liu J, Puolanne E, Schwartzkopf M, Arner A. Altered sarcomeric structure and function in woody breast myopathy of avian pectoralis major muscle. Front Physiol. 2020;11:287. https://doi.org/10.3389/fphys.2020.00287.
Kang H, Zhao D, **ang H, Li J, Zhao G, Li H. Large-scale transcriptome sequencing in broiler chickens to identify candidate genes for breast muscle weight and intramuscular fat content. Genet Sel Evol. 2021;53:66. https://doi.org/10.1186/s12711-021-00656-9.
Rashid Z, Mirani ZA, Zehra S, Gilani S, Ashraf A, Azhar A, et al. Enhanced modulation of gut microbial dynamics affecting body weight in birds triggered by natural growth promoters administered in conventional feed. Saudi J Biol Sci. 2020;27(10):2747–55. https://doi.org/10.1016/j.sjbs.2020.06.027.
Chen B, Yu J, Guo L, Byers MS, Wang Z, Chen X, et al. Circular RNA circHIPK3 promotes the proliferation and differentiation of chicken myoblast cells by sponging miR-30a-3p. Cells-Basel. 2019;8(2):177. https://doi.org/10.3390/cells8020177.
Suzuki S, Kobayashi M, Murai A, Tsudzuki M, Ishikawa A. Characterization of growth, fat deposition, and lipid metabolism-related gene expression in lean and obese meat-type chickens. J Poult Sci. 2019;56(2):101–11. https://doi.org/10.2141/jpsa.0180064.
Aarts HJ, Van Der Hulst-Van AM, Beuving G, Leenstra FR. Variations in endogenous viral gene patterns in white leghorn, medium heavy, white plymouth rock, and cornish chickens. Poult Sci. 1991;70(6):1281–6. https://doi.org/10.3382/ps.0701281.
Moreira G, Boschiero C, Cesar A, Reecy JM, Godoy TF, Trevisoli PA, et al. A genome-wide association study reveals novel genomic regions and positional candidate genes for fat deposition in broiler chickens. BMC Genomics. 2018;19:374. https://doi.org/10.1186/s12864-018-4779-6.
Buzala M, Janicki B. Review: effects of different growth rates in broiler breeder and layer hens on some productive traits. Poult Sci. 2016;95(9):2151–9. https://doi.org/10.3382/ps/pew173.
Nie Y, Sato Y, Wang C, Yue F, Kuang S, Gavin TP. Impaired exercise tolerance, mitochondrial biogenesis, and muscle fiber maintenance in miR-133a-deficient mice. Faseb J. 2016;30(11):3745–58. https://doi.org/10.1096/fj.201600529R.
Lowey S, Bretton V, Joel PB, Trybus KM, Gulick J, Robbins J, et al. Hypertrophic cardiomyopathy R403Q mutation in rabbit β-myosin reduces contractile function at the molecular and myofibrillar levels. Proc Natl Acad Sci U S A. 2018;115(44):11238–43. https://doi.org/10.1073/pnas.1802967115.
Hou L, Xu J, Li H, Ou J, Jiao Y, Hu C, et al. MiR-34c represses muscle development by forming a regulatory loop with Notch1. Sci Rep. 2017;7(1):9346. https://doi.org/10.1038/s41598-017-09688-y.
Gao X, Zhao Y, Stemmer-Rachamimov AO, Liu H, Huang P, Chin S, et al. Anti-VEGF treatment improves neurological function and augments radiation response in NF2 schwannoma model. Proc Natl Acad Sci U S A. 2015;112(47):14676–81. https://doi.org/10.1073/pnas.1512570112.
Ye C, Zhang D, Zhao L, Li Y, Yao X, Wang H, et al. CaMKK2 suppresses muscle regeneration through the inhibition of myoblast proliferation and differentiation. Int J Mol Sci. 2016;17(10):1695. https://doi.org/10.3390/ijms17101695.
Sun H, Jiang R, Xu S, Zhang Z, Xu G, Zheng J, et al. Transcriptome responses to heat stress in hypothalamus of a meat-type chicken. J Anim Sci Biotechnol. 2015;61:6. https://doi.org/10.1186/s40104-015-0003-6.
Dewulf M, Köster DV, Sinha B, Viaris DLC, Chambon V, Bigot A, et al. Dystrophy-associated caveolin-3 mutations reveal that caveolae couple IL6/STAT3 signaling with mechanosensing in human muscle cells. Nat Commun. 2019;10:1974. https://doi.org/10.1038/s41467-019-09405-5.
Yang Y, Fan X, Yan J, Chen M, Zhu M, Tang Y, et al. A comprehensive epigenome atlas reveals DNA methylation regulating skeletal muscle development. Nucleic Acids Res. 2021;49(3):1313–29. https://doi.org/10.1093/nar/gkaa1203.
Tan X, Liu R, Li W, Zheng M, Zhu D, Liu D, et al. Assessment the effect of genomic selection and detection of selective signature in broilers. Poult Sci. 2022;101(6):101856. https://doi.org/10.1016/j.psj.2022.101856.
Antón-García P, Haghighi EB, Rose K, Vladimirov G, Boerries M, Hecht A. TGFβ1-induced EMT in the MCF10A mammary epithelial cell line model is executed independently of SNAIL1 and ZEB1 but relies on JUNB-coordinated transcriptional regulation. Cancers (Basel). 2023;15(2):558. https://doi.org/10.3390/cancers15020558.
Li J, Sun C, Zheng J, Li J, Yi G, Yang N. Time-course transcriptional and chromatin accessibility profiling reveals genes associated with asymmetrical gonadal development in chicken embryos. Front Cell Dev Biol. 2022;10:832132. https://doi.org/10.3389/fcell.2022.832132.
Reed A, Ware T, Li H, Fernando BJ, Cravatt BF. TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat Chem Biol. 2023;19:378–88. https://doi.org/10.1038/s41589-022-01253-7.
Wang D, Qin P, Zhang K, Wang Y, Guo Y, Cheng Z, et al. Integrated LC/MS-based lipidomics and transcriptomics analyses revealed lipid composition heterogeneity between pectoralis intramuscular fat and abdominal fat and its regulatory mechanism in chicken. Food Res Int. 2023;172:113083. https://doi.org/10.1016/j.foodres.2023.113083.
Luo W, Lin Z, Chen J, Chen G, Zhang S, Liu M, et al. TMEM182 interacts with integrin beta 1 and regulates myoblast differentiation and muscle regeneration. J Cachexia Sarcopenia Muscle. 2021;12(6):1704–23. https://doi.org/10.1002/jcsm.12767.
Suresh R, Diaz RJ. The remodelling of actin composition as a hallmark of cancer. Transl Oncol. 2021;14(6):101051. https://doi.org/10.1016/j.tranon.2021.101051.
Ciecierska A, Motyl T, Sadkowski T. Transcriptomic profile of primary culture of skeletal muscle cells isolated from semitendinosus muscle of beef and dairy bulls. Int J Mol Sci. 2020;21(13):4794. https://doi.org/10.3390/ijms21134794.
Wang C, Lei B, Liu Y. An analysis of a transposable element expression atlas during 27 developmental stages in porcine skeletal muscle: unveiling molecular insights into pork production traits. Animals (Basel). 2023;13(22):3581. https://doi.org/10.3390/ani13223581.
Hicks MR, Saleh KK, Clock B, Gibbs DE, Yang M, Younesi S, et al. Regenerating human skeletal muscle forms an emerging niche in vivo to support PAX7 cells. Nat Cell Biol. 2023;25(12):1758–73. https://doi.org/10.1038/s41556-023-01271-0.
Chen P, **g H, **ong M, Zhang Q, Lin D, Ren D, et al. Spine impairment in mice high-expressing neuregulin 1 due to LIMK1 activation. Cell Death Dis. 2021;12(4):403. https://doi.org/10.1038/s41419-021-03687-8.
Morano M, Ronchi G, Nicolò V, Fornasari BE, Crosio A, Perroteau I, et al. Modulation of the Neuregulin 1/ErbB system after skeletal muscle denervation and reinnervation. Sci Rep. 2018;8:5047. https://doi.org/10.1038/s41598-018-23454-8.
Rossi M, Hall CM, Bouvier R, Collardeau-Frachon S, Le Breton F, Bucourt M, et al. Radiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis. Pediatr Radiol. 2015;45(7):965–76. https://doi.org/10.1007/s00247-014-3257-9.
Tsushima H, Tang YJ, Puviindran V, Hsu SC, Nadesan P, Yu C, et al. Intracellular biosynthesis of lipids and cholesterol by Scap and Insig in mesenchymal cells regulates long bone growth and chondrocyte homeostasis. Development. 2018;145(13):dev162396. https://doi.org/10.1242/dev.162396.
Chen W, Xu J, Wu Y, Liang B, Yan M, Sun C, et al. The potential role and mechanism of circRNA/miRNA axis in cholesterol synthesis. Int J Biol Sci. 2023;19(9):2879–96. https://doi.org/10.7150/ijbs.84994.
Rienhoff HJ, Yeo CY, Morissette R, Khrebtukova I, Melnick J, Luo S, et al. A mutation in TGFB3 associated with a syndrome of low muscle mass, growth retardation, distal arthrogryposis and clinical features overlap** with marfan and loeys-dietz syndrome. Am J Med Genet A. 2013;161A(8):2040–6. https://doi.org/10.1002/ajmg.a.36056.
Lu Y, Chen S, Yang N. Expression and methylation of FGF2, TGF-β and their downstream mediators during different developmental stages of leg muscles in chicken. PLoS ONE. 2013;8(11):e79495. https://doi.org/10.1371/journal.pone.0079495.
Almada AE, Horwitz N, Price FD, Gonzalez AE, Ko M, Bolukbasi OV, et al. Fos licenses early events in stem cell activation driving skeletal muscle regeneration. Cell Rep. 2021;34(4):108656. https://doi.org/10.1016/j.celrep.2020.108656.
Kim CJ, Singh C, Kaczmarek M, O’Donnell M, Lee C, Dimagno K, et al. Mustn1 ablation in skeletal muscle results in functional alterations. FASEB Bioadv. 2023;5(12):541–57. https://doi.org/10.1096/fba.2023-00082.
Krause MP, Moradi J, Coleman SK, D’Souza DM, Liu C, Kronenberg MS, et al. A novel GFP reporter mouse reveals Mustn1 expression in adult regenerating skeletal muscle, activated satellite cells and differentiating myoblasts. Acta Physiol (Oxf). 2013;208(2):180–90. https://doi.org/10.1111/apha.12099.
Liu C, Gersch RP, Hawke TJ, Hadjiargyrou M. Silencing of Mustn1 inhibits myogenic fusion and differentiation. Am J Physiol Cell Physiol. 2010;298(5):C1100–8. https://doi.org/10.1152/ajpcell.00553.2009.
Li Y, Yuan J, Chen F, Zhang S, Zhao Y, Chen X, et al. Long noncoding RNA SAM promotes myoblast proliferation through stabilizing Sugt1 and facilitating kinetochore assembly. Nat Commun. 2020;11:2725. https://doi.org/10.1038/s41467-020-16553-6.
Vila MC, Rayavarapu S, Hogarth MW, Van der Meulen JH, Horn A, Defour A, et al. Mitochondria mediate cell membrane repair and contribute to duchenne muscular dystrophy. Cell Death Differ. 2017;24(2):330–42. https://doi.org/10.1038/cdd.2016.127.
Acknowledgements
We thank Bei**g Huadu Yukou Poultry Industry Co., Ltd. for providing the experimental broiler chickens and Professor **anyao Li of Shandong Agricultural University for the assistance in samples collection.
Funding
This work was supported by the National Key Research and Development Program of China (2022YFF1000204), the National Natural Science Foundation of China (32102535) and the Key Research and Development Program of Hainan province (ZDYF2023XDNY036).
Author information
Authors and Affiliations
Contributions
NY and CW conceived and designed the project. SG, QH, CW, YJ and CS performed the experiments and collected samples. SG analyzed the data and wrote the manuscript. NY and CW revised the manuscript. All authors read and approved the final draft.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All experimental procedures associated with the chickens used in this study were carried out in accordance with the guidelines established by the Ministry of Science and Technology (Bei**g, China). Ethical approval was obtained from the Animal Welfare Committee of China Agricultural University (AW71802202-1-2) and adhered to the protocol outlined in the “Guide for Care and Use of Laboratory Animals”.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary Information
Additional file 1: Table S1.
Experimental sample size of each analysis. Table S2. Summary of sequencing quality and reads alignment statistics of RNA-seq data. Table S3. Summary of sequencing quality and reads alignment statistics of ATAC-seq data. Table S4. Growth performance of CC and RR from DOH to D42. Table S5. Differentially expressed genes profiles between D21 and D42 of CC. Table S6. Differentially expressed genes profiles between D21 and D42 of RR. Table S7. Differentially expressed genes profiles between CC and RR at D21. Table S8. Differentially expressed genes profiles between CC and RR at D42. Table S9. Differential accessible regions profiles between D21 and D42 of CC. Table S10. Differential accessible regions profiles between D21 and D42 of RR. Table S11. Differential accessible regions profiles between CC and RR at D21. Table S12. Differential accessible regions profiles between CC and RR at D42. Table S13. The DEGs carrying with DARs.
Additional file 2:
Fig. S1. KEGG pathway enrichment analysis on the differential gene. a Function analysis on differential genes between CC and RR at D21 and D42. b Function analysis on differential genes between D21 and D42 of CC and RR. Fig. S2. Chromatin accessibility analysis of CC and RR at D21 and D42. a–c Fragment insert size of CC-D42, RR-D21 and RR-D42 groups. d Number of peaks in the four groups. e Heatmaps of differential accessible regions (DARs). f The relationship between differentially expressed genes (DEGs) and DARs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Gu, S., Huang, Q., Jie, Y. et al. Transcriptomic and epigenomic landscapes of muscle growth during the postnatal period of broilers. J Animal Sci Biotechnol 15, 91 (2024). https://doi.org/10.1186/s40104-024-01049-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40104-024-01049-w