
Abstract
Background
Hemophilia B is a rare inherited genetic bleeding disorder caused by a deficiency or lack of coagulation factor IX, the gene for which (F9) is located on the X chromosome. Hemophilia B is currently incurable and the standard treatment is coagulation factor replacement therapy. Although gene therapy has the potential to cure hemophilia, significant barriers are still needed to be overcome, e.g., off-target effects and immunoreactivity, so new approaches must be explored. Nonsense mutations account for 8% of all the hemophilia B mutation types and can result in the development of coagulation factor inhibitors. In this study, CRISPR/Cas9 technology was used to construct a mouse embryonic stem cell model with a hemophilia B nonsense mutation (F9 c.223C > T) in humans to investigate the pathogenesis and treatment of nonsense mutations in hemophilia B.
Methods
First, a donor plasmid with a mutation (F9 c.223 C > T) and sgRNAs were constructed. Second, both the donor plasmid and the px330-sgRNA were electroporated into mouse embryonic stem cell, and the mutant cells were then screened using puromycin and red fluorescence. Third, the mutant cell lines were tested for pluripotency and the ability to differentiate into three layers. Finally, the effect of mutation on gene function was studied in the differentiation system.
Results
The mutant vector and effective sgRNA were constructed, and the mutant cell line was screened. This mutant cell line exhibited pluripotency and the ability to differentiate into three layers. This point mutation affects F9 expression at both the RNA and protein levels in the differentiation system.
Conclusions
The mutant cell line obtained in the current study had a single-base mutation rather than a base deletion or insertion in the exon, which is more similar to clinical cases. In addition, the mutant has the characteristics of mouse embryonic stem cells, and this point mutation affects F9 gene transcription and translation, which can be used as a disease model for studying the pathogenesis and treatment of hemophilia at the stem cell level.
Similar content being viewed by others

Background
The clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene-editing system has become a powerful genome-editing tool in the field of molecular biology since its discovery. When introduced into cells, the Cas9 protease is directed to the target genome sequence by a specific synthetic sgRNA and subsequently breaks both DNA strands. The resulting double-strand breaks are then repaired by either the error-prone nonhomologous end-joining or homology-directed repair, resulting in genomic DNA knock-out or knock-in [1, 2].
Hemophilia B is a rare X-linked bleeding disorder caused by a deficiency or lack of coagulation factor IX (FIX). The F9 gene is located on the X chromosome. Hemophilia B is recessively inherited and is predominantly expressed in males because males have only one X chromosome [3, 4]. The type of causative mutation determines the severity degree of hemophilia, which can be classified into three levels based on the residual FVIII or FIX activity as follows: severe (< 1 international unit [IU]/dl), moderate (1–5 IU/dl), or mild (6 to < 40 IU/dl) [5, 6]. The standard therapy for severe patients in developed countries requires prophylactic treatment with replacement factors, while patients with severe conditions in develo** countries are treated mainly on demand [7]. The most serious complication of hemophilia B replacement therapy is the development of neutralizing antibodies (inhibitors) against FIX. At high titers, these inhibitors may reduce the efficacy of replacement factors or even render replacement therapy completely ineffective, increasing the burden on family and society [8]. Although gene therapy has the potential to cure this disease, factors (e.g., immunogenicity and off-target effects) remain unresolved.
Nonsense mutations may increase the risk for the development of coagulation factor inhibitors [9]. The hemophilia B mutation database (http://www.factorix.org/) revealed 87 different forms of nonsense mutations, accounting for 8% of all hemophilia B mutation types. An extensive meta-analysis study based on the Human Gene Mutation Database showed that 11% of all genetic disease-causing mutations are nonsense mutations resulting in the translation of premature termination codons (PTCs) [10]. Therefore, a new therapeutic strategy for hemophilia with a nonsense mutation should be developed.
Truncated proteins are produced when PTCs are introduced into nonsense mutations, resulting in gene inactivation. Nonsense mutations may also reduce messenger RNA levels due to the rapid mRNA turnover caused by the nonsense-mediated mRNA decay pathway [11]. Aminoglycosides and several small molecular compounds can bind to the ribosome decoding center, reducing the accuracy of codon and anticodon pairing, promoting PTC binding to near-cognate aminoacyl tRNA, and decoding PTC into a meaningful codon. Protein synthesis produces a constant number of functional full-length proteins, which is used in translational bypass therapy for nonsense mutations [12, 13].
The effect of read-through therapy is related to the nonsense mutant gene mRNA stability. A strong read-through effect can be acquired only when the mRNA (PTC-mRNA)-containing PTC is stable [14, 15]. New compounds that can promote the premature stop codon read-through in nonsense mutations have been recently developed. For instance, ELX-02, which is designed to treat genetic diseases caused by nonsense mutations (e.g., cystic fibrosis and cystinosis) is currently being studied in clinical trials. These synthetic small molecular drugs have shed light on the treatment of hemophilia B. To investigate read-through treatment, the present study aimed to generate an F9 nonsense mutation mouse embryonic stem cell (mESC) that is closer to clinical cases without gene deletion or insertion but only a single-base mutation. This cell model is also useful for studying the mechanism and treatment of hemophilia B.
Since a nonsense mutation of F9 c.223C > T (p.R75X) was detected in a patient with hemophilia B at the hospital of the current study and the mouse with this mutation encodes the same amino acid as humans, an mESC with this mutation was constructed.
Methods
Plasmid construction
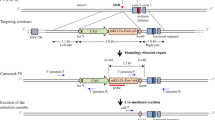
The primers were designed to amplify and purify the left and right arms, as well as the Loxp-PGK-puro-p2A-Mcherry-Loxp via polymerase chain reaction (PCR), with the mutation point on the left arm (F9 c.223C > T). The donor plasmids (PGGA-M. LA-LOXP-PGK-Puro-P2A-Mcherry-LOXP-RA) were assembled using the Golden Gate method (E1601 NEB, BsaI-HFV2). The sgRNA was designed on the website [16] (https://zlab.bio/guide-design-resources) and inserted into the pX330 plasmid, which targets the second intron of the F9 gene. The vector sequences are shown in Additional file 2: Text 1, and the primers’ sequences are listed in Table 1.
Cell culture
The E14 mESCs were cultured on gelatin-coated plates in Dulbecco’s Modified Eagle’s Medium–high glucose (DMEM, HyClone, Logan, UT, USA) supplemented with 20% fetal bovine serum (FBS, Gibco, ThermoFisher Scientific, Waltham, MA, USA), nonessential amino acids (1 mM, Gibco), GlutaMAX (1×, Gibco), sodium pyruvate (1 mM, Gibco), penicillin (50 units/mL), streptomycin (50 μg/mL, Hyclone), β-mercaptoethanol (0.1 mM), leukemia inhibitory factor (1000 U/mL), and 2i inhibitors (3 μM CHIR99021 and 1 μM PD0325901). The plates were incubated at 37 °C in a humidified incubator with 5% CO2, and the media was changed daily. The cells were detached using a 0.25% trypsin–EDTA solution (Gibco) at a 1:10 split ratio when the confluence reached 70%.
Generation of F9 mutation in mESCs
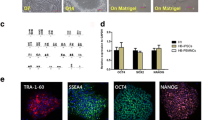
Using the Ltd.P3 Primary Cell 4D-NucleofectorTM X Kit L (V4XP-3012, Lonza Group AG, Basel, Switzerland), 1 × 106 mESCs were electroporated with a mixture of 6 and 2 μg donor and sgRNA-pX330 plasmids, respectively. After electroporation, the cells were cultured in a 10-cm culture dish for 12 h and screened with 1 μg/mL puromycin (Gibco) for two passages, refreshing the media every day. When the confluence reached 70%, individual clones with higher fluorescence were selected for further screening on 48-well gelatin-coated plates (one clone per well). The genomic DNA was extracted using the TIANamp genomic DNA kit (Tiangen Biotech Co., Ltd., Bei**g, China). The individual clones were further verified using “F9-F-flanked” and “F9-R-flanked” primers (Fig. 1a), which confirmed that the modification in F9 intron 2 was caused by CRISPR/Cas9-mediated donor insertion.

Gene mutation using CRISPR/Cas9 technology and screening of mutant cells. a Gene pattern diagram. b The experimental procedure. c (I) Editing rates of the three sgRNA, the binding position of each sgRNA is different. The numbers represent the distance from the downstream of the mutation site to that of sgRNA, and c (II) after sgRNA3 and donor transfection, the mutant cell line was screened out (E14-mF9-27#). d After donor and sgRNA3 transfection, partial PCR results of flanking primer (PCR product) was 1449 bp. 27# (E14-mF9-27#), M DNA marker, C1 represents negative control, and C2 represents near-positive control
Immunocytochemistry
The cells were washed twice with phosphate-buffered saline (PBS) and fixed in paraformaldehyde (4%) for 15 min at room temperature. The cells were then washed twice with PBS, permeabilized in PBS containing 0.3% Triton X-100 for 10 min, washed thrice with PBS, then blocked in 5% bovine serum albumin (BSA) at 37 °C for 1 h. The cells were then incubated overnight at 4 °C with rabbit anti-NANOG and mouse anti-OCT4 diluted in 1% BSA. The cells were then washed thrice with PBS for 5 min each to remove any unbound primary antibodies and incubated for 1.5 h with fluorescein-conjugated antirabbit and antimouse secondary antibodies (both 1:400) in 1% BSA. The cells were then washed thrice with PBS, stained with 4′,6-diamidino-2-phenylindole (1 μg/mL for 10 min), washed three more times with PBS, and imaged using a confocal laser scanning microscope (Zeiss LSM 710). The antibodies used in the present study are listed in Table 2.
Embryoid bodies differentiation
For embryoid bodies (EB) formation, the cells were cultured in hanging drops (1,000/2,000 cells per drop) for 4 days in a DMEM (HyClone) differentiation medium containing 20% FBS (Gibco), nonessential amino acids (1 mM, Gibco), GlutaMAX (1 × , Gibco), sodium pyruvate (1 mM, Gibco), penicillin (50 units/mL), streptomycin (50 μg/mL, HyClone), and β-mercaptoethanol (0.1 mM). The EBs were then transferred into 60-mm Petri dishes and cultured in a differentiation medium for another 2–5 days. The expression of the three germ maker genes was detected on total RNA using quantitative PCR.
Hepatocyte-like cells differentiation
The cells were cultured for 6 days on 24-well gelatin-coated plates (0.1%) at a density of 2 × 104 cells/well in a differentiation medium containing 2.5 mmol/L sodium butyrate (Sigma-Aldrich, St. Louis, MO, USA), 10 ng/mL basic fibroblast growth factor (R&D Systems, Minneapolis, MN, USA), and 10 ng/mL bone morphogenetic protein 4 (R&D Systems). The medium was then replaced with a differentiation medium containing 20 ng/mL hepatocyte growth factor (R&D Systems) and incubated for another 6 days. During the last 3 days of culture, 0.1 μmol/L dexamethasone (Sigma-Aldrich) and 10 ng/mL oncostatin M (R&D Systems) was added to the medium (Fig. 3a) [17].
RNA isolation and real-time qPCR
Total RNA was extracted using the EZ-press RNA purification kit (EZBioscience, Roseville, MN, USA), and cDNAs were synthesized using the HiScript® III RT SuperMix for qPCR (Nan**g Vazyme Biotech Co., Nan**g, China). The specific gene expression was assayed using real-time qPCR (RT-qPCR) with 2× RealStar Green Power Mixture (Nan**g Vazyme Biotech Co.) on CFX Connect (Bio-Rad Laboratories, Hercules, CA, USA) with the following thermal profile: 95 °C for 10 min, followed by 40 cycles of 95 °C for 10 s, 60 °C for 10 s, and 72 °C for 20 s. The primers’ sequences used in RT-qPCR assays are listed in Table 1.
Western blotting
The cells were lysed with radioimmunoprecipitation assay lysate and centrifuged at 12,000 RPM for 10 min at 4℃. The supernatant was collected and the protein concentration was measured. The protein sample (20 µg) was added to a 5× loading buffer, boiled at 100℃ for 10 min, and then electrophoresed in Tris–glycine buffer. The samples were then wet transferred onto a nitrocellulose membrane and blocked in 5% nonfat milk for 60 min. The membrane was washed thrice with Tris-buffered saline with Tween (TBST) for 5 min each and incubated with the primary antibody overnight at 4℃. After washing thrice with TBST for 5 min each, the membrane was incubated with the corresponding secondary antibody at room temperature for 1–2 h and washed thrice with TBST for 10 min each. Enhanced chemiluminescence solutions A and B were mixed with a volume ratio of 1:1 and dropped onto the surface of ECL in a darkroom. Exposure was made after 1 min of incubation. The antibodies used in this assay are listed in Table 2.
Karyoty**
The karyoty** analysis of the mutant cell line was conducted by Guangzhou Chunshui Bioscience & Technology Co. (Guangzhou, China). Twenty metaphase spreads were randomly selected for analysis, and the results revealed that the cell line generated in this study had a normal karyotype with no detectable chromosomal abnormalities.
Cre-RNA synthesis
The Cre-RNA was synthesized using the TR101–T7 high-yield RNA transcription kit (Vazyme) following the manufacturer’s instructions and stored separately (400 ng per tube) after synthesis at − 80℃.
Short tandem repeat analysis
The short tandem repeat (STR) analysis (Shanghai QiDa Biotechnology, Shanghai, China) revealed that the E14-F9 mutant cell line was identical to the parent cell line. The STR analysis is shown in the word document Additional file 1: “STR Analysis.”
Results
Vectors construction and sgRNA integration efficiency rate
The donor plasmid with the mutation was assembled according to the Golden Gate method. The screening purinomycin gene and red fluorescence are located between the homologous left and right arms. Loxp sequences pointing in the same direction were inserted into both ends of the screening gene (Fig. 1a), and sgRNA at different positions was selected (Fig. 1b). After plasmid construction, mouse embryonic stem cells were electrocuted and then screened in a pure medium. On the second day following the electrocution, monoclonal cells with red fluorescence were manually selected for further culture, and PCR detection was performed after amplification (Fig. 1b). The sgRNA1 sequence is as follows: CACCG TTTTTGAGTAGTACATACAC (forward) and AAAC GTGTATGTACTACTCAAAAAC (reverse). The sgRNA2 sequence is as follows: CACCGAAACTAGAAGAGTTTGTTCG (forward) and AAACCGAACAAACTCTTCTAGTTTC (reverse). The sgRNA3 sequence is as follows: CACCGAAAGGAGTGTGGAACTAGAG (forward) and AAACCTCTAGTTCCACACTCCTTTC (reverse).
sgRNA1 was off target by the detection of flanking primers. The sgRNA2 editing rate was 6.26%, with three of the 48 selected clones exhibiting the intended homologous recombination, and all three clones had mutated target sites. However, base deletion or insertion occurred at the Cas9 cleavage site because the sgRNA was located on the exon. The second Cas9 cleavage can be avoided by using the synonym mutation in the donor plasmid’s protospacer adjacent motif (PAM) region, but the PAM region of sgRNA2 only recodes glutamate. The sgRNA3 editing rate was 15.27%, with 11 of the 72 selected clones exhibiting the intended homologous recombination, which was greater than the sgRNA2 editing rate. However, given the distance between sgRNA3 and the mutation site, only one of the 11 cell clones (E14-mF9-27#) had the target-site mutation. Two bases “TA” were inserted into the intron at the site of Cas9 cutting without affecting gene expression (Fig. 1c-ii). Based on the Zhang Laboratory website [16], four off-target sites were verified, and no off-target event was detected with this study’s designed sgRNA3.
Obtained mutant cell line
The mouse embryonic stem cells were transferred by donor and sgRNA3 and then cultured in a purinomycin environment. After passage, the surviving monoclonal cells with red fluorescence were manually selected for further PCR detection and Sanger sequencing, and the mutant cell line E14-mF9-27 # (E14-F9 C.223 C > T) was acquired (Fig. 1d). The acquired mutant was then electrotransfected with Cre-RNA to cut off the Loxp-PGK-puro-p2A-Mcherry-Loxp sequence from the genome, leaving only one Loxp sequence [Full size image
The effect of point mutation on F9 gene function in differentiation system
The effect of this point mutation on gene function in the differentiation system was investigated. The F9 gene was found to be mostly expressed in the mature liver with minimal expression in the fetal liver and no expression in embryonic stem cells [19]. Previous studies have demonstrated that induced hepatocyte-like cells can express coagulation factors VIII and IX [17, 20]. Embryonic stem cells were differentiated into hepatocyte-like cells following the previously described protocol [17] (Fig. 3a) and observed cell morphological changes during differentiation. The cells change from a round stem cell shape to a fusiform-defined endoderm shape and eventually differentiate into polygonal hepatoid cells. No obvious morphological difference was noted between mutant (Mu-Hep) and nonmutant (WT-hep) groups, but the cell morphology of the naturally differentiated group (WT-N) was different and irregular (Fig. 3b). On day 15 of differentiation, albumin (ALB), a specific markers of hepatocyte-like cells, could be detected in Mu-Hep and WT-Hep, but F9 expression was detected only in WT-Hep (Fig. 4a–c). In addition, point mutations do not affect hepatocyte-like cell differentiation but affect F9 gene transcription and translation, similar to the human patient with this mutation, but the patient was a severe hemophiliac.

Differentiation of hepatocyte-like cells. a A diagram of differentiation patterns. b Morphological changes of cells during differentiation

Marker detection in hepatoid cells. a RNA expression levels of various markers at days 0, 6, and 15 of differentiation, b immunofluorescence staining after the 15th day of differentiation, and c protein expression on the 15th day of differentiation
