
Abstract
Background
Aggregation of the amyloid-β (Aβ) peptide in the brain is one of the key pathological events in Alzheimer’s disease (AD). Reducing Aβ levels in the brain by enhancing its degradation is one possible strategy to develop new therapies for AD. Neprilysin (NEP) is a membrane-bound metallopeptidase and one of the major Aβ-degrading enzymes. The secreted soluble form of NEP (sNEP) has been previously suggested as a potential protein-therapy degrading Aβ in AD. However, similar to other large molecules, peripherally administered sNEP is unable to reach the brain due to the presence of the blood–brain barrier (BBB).
Methods
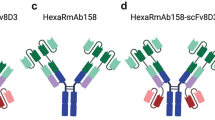
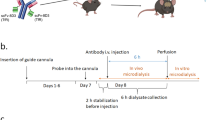
To provide transcytosis across the BBB, we recombinantly fused the TfR binding moiety (scFv8D3) to either sNEP or a previously described variant of NEP (muNEP) suggested to have higher degradation efficiency of Aβ compared to other NEP substrates, but not per se to degrade Aβ more efficiently. To provide long blood half-life, an Fc-based antibody fragment (scFc) was added to the designs, forming sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3. The ability of the mentioned recombinant proteins to degrade Aβ was first evaluated in vitro using synthetic Aβ peptides followed by sandwich ELISA. For the in vivo studies, a single injection of 125-iodine-labelled sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3 was intravenously administered to a tg-ArcSwe mouse model of AD, using scFc-scFv8D3 protein that lacks NEP as a negative control. Different ELISA setups were applied to quantify Aβ concentration of different conformations, both in brain tissues and blood samples.
Results
When tested in vitro, sNEP-scFc-scFv8D3 retained sNEP enzymatic activity in degrading Aβ and both constructs efficiently degraded arctic Aβ. When intravenously injected, sNEP-scFc-scFv8D3 demonstrated 20 times higher brain uptake compared to sNEP. Both scFv8D3-fused NEP proteins significantly reduced aggregated Aβ levels in the blood of tg-ArcSwe mice, a transgenic mouse model of AD, following a single intravenous injection. In the brain, monomeric and oligomeric Aβ were significantly reduced. Both scFv8D3-fused NEP proteins displayed a fast clearance from the brain.
Conclusion
A one-time injection of a BBB-penetrating NEP shows the potential to reduce, the likely most toxic, Aβ oligomers in the brain in addition to monomers. Also, Aβ aggregates in the blood were reduced.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder and the most common form of dementia. It is characterized by the extracellular deposition of amyloid plaques and the formation of intracellular neurofibrillary tangles comprised of tau protein [1]. Amyloid plaques are mainly comprised of amyloid-beta (Aβ) peptides. Aβ is formed by enzymatic digestion of Aβ precursor protein (APP), giving rise to Aβ peptides of different lengths [2]. The 40 amino acid long peptide (Aβ40) is the most abundant Aβ form in the human brain followed by Aβ42, an isoform that is more prone to aggregate. Under physiological condition, Aβ exists as an unfolded monomer with studies demonstrating possible physiological roles of this peptide in neurogenesis and lipid homeostasis [3,4,5,6]. However, during AD, partially folded Aβ monomers start to form aggregates of different sizes and solubility, ranging from soluble oligomers with hairpins and protofibrils to insoluble fibrils and finally amyloid plaques [7, 8]. Soluble aggregates (oligomers and protofibrils) have been demonstrated to correlate with AD progression and are suggested to be the most neurotoxic species of Aβ [9,10,11,12,17, 38, 39, 61] was recombinantly linked to soluble NEP (sNEP) and an Fc-region fragment (scFc) of mouse IgG2c antibody, forming the final protein drug (sNEP-scFc-scFv8D3). In addition to the mentioned protein, we designed another recombinant protein (muNEP-scFc-scFv8D3) that was based on a mutated variant of NEP displaying lower efficiency in degrading other NEP substrates compared to Aβ [44].
Following purification, the two NEP proteins were present mainly in the monomeric form with a strong band in SDS-PAGE at around 160 kDa (Fig. 2A). However, SDS-PAGE analysis demonstrated some weaker bands that were bigger than 160 kDa, suggesting the formation of protein multimers. A previous study has demonstrated the dimerization of naturally occurring NEP [62], which could suggest that the formation of sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3 multimers is related to the presence of NEP in these designs. Nevertheless, the possibility that the other domains such as scFc contributed to the formation of these multimers cannot be entirely excluded. A rather rapid degradation of the NEP constructs could be detected in vivo, a possible reason for this could be the propensity to form multimers which often leads to more rapid degradation.
The ability of sNEP-scFc-scFv8D3 to degrade Aβ was first assessed in vitro using sNEP as the positive control. In our designs, the N-terminal of sNEP is fused to other protein domains (scFc-scFv8D3). Therefore, we wanted to evaluate whether such protein engineering might interfere with the ability of NEP to degrade Aβ. sNEP-scFc-scFv8D3 preserved NEP properties in binding to and degrading Aβ monomers in vitro (Figs. 3 and 4). These results indicate that the recombinant addition of scFc and scFv8D3 on the N-terminal end of NEP is a suitable strategy to develop NEP-based protein drugs with long half-life and capability to cross BBB, respectively.
sNEP-scFc-scFv8D3 could, similar to sNEP, significantly degrade Aβ1-40 and Aβ1-42 monomers in vitro (Fig. 5) both at the 1:1 and 5:1 ratios. Surprisingly, the recombinant protein based on the mutated NEP protein (muNEP-scFc-scFv8D3) was not able to degrade wt-Aβ at a 5:1 ratio, where the concentration of Aβ was higher, 2.5 μM. Degradation was evident only when a 1:1 ratio of Aβ and NEP proteins was used and with a 0.5-μM concentration of Aβ (Fig. 4). The higher the concentration of Aβ, the faster the aggregation is a well-known fact. It is possible that the concentration of 2.5 μM was high enough to start aggregation already before the first time point analysed and that the muNEP is worse at degrading aggregates than sNEP. muNEP has been previously reported to exert a 20 times higher Aβ degradation capacity compared to wild-type NEP but this was at much higher concentrations of Aβ [44] and a much lower ratio than we had at other concentrations it seemed from the data that it was less effective. Wild-type NEP (sNEP in our study) has the capacity to degrade Aβ at several cleavage sites between amino acids 1 to 35. However, muNEP preferentially degrades Aβ at position 20–21 located at the mid-region of Aβ [44]. This region becomes gradually hidden with the formation of aggregates, further suggesting the inability of muNEP to degrade aggregated forms Aβ.
The in vitro Aβ degradation results (Fig. 4) demonstrated the ability of sNEP-scFc-scFv8D3 to efficiently degrade both Aβ1-40 and Aβ1-42 monomers. Interestingly, sNEP-scFc-scFv8D3 could degrade Aβ1-40 faster than Aβ1-42 at high Aβ concentrations (Fig. 4A–C). NEP cleavage sites are located within the first 35 amino acids of Aβ [63], meaning that the enzyme should have an equal degradation efficiency on both Aβ isoforms. NEP is more efficient in degrading monomers, and since Aβ1-42 is more prone to aggregate, it is more likely that this isoform started to aggregate during the experiment, hence slower degradation of this isoform compared to Aβ1-40. Our results are in agreement with [64], demonstrating NEP ability to degrade synthetic Aβ1-40 and Aβ1-42 peptides in vitro. Similar to our results, the mentioned study demonstrated NEP ability to degrade Aβ1-40 faster compared to Aβ1-42. On the contrary, we have recently demonstrated a selective degradation of Aβ42 (but not Aβ40) in the hippocampus of AD mice mediated by enhanced NEP expression following treatment with a brain-penetrating somatostatin peptide [17]. However, our previous study demonstrated the degradation efficiency of membrane-bound NEP, while the current study is based on the use of the soluble part of NEP (sNEP). These studies suggest NEP-mediated degradation of Aβ to be dependent on several factors such as the availability and concentration of Aβ, its aggregation state and the form of NEP (membrane-bound or soluble).
A bit to the contrary of the discussion above, both NEP constructs degraded the fast-aggregating arctic Aβ the best in the conditions used. A possible reason for this can be that arctic Aβ is known to form protofibrils but less of insoluble aggregates [46]. The structure of these protofibrils has not been determined, but they possibly retain the beta hairpin that is also present in oligomers and the first thing that forms when monomers start to aggregate. This hair pin converts to larger beta sheets in the larger aggregates. In the modelling of NEP binding site to Aβ, it seems like this hair pin still could be accessible while the beta sheets of the aggregates would not be. That is, it is possible that NEP can degrade the aggregates formed by arctic Aβ, but not other types of aggregates. That this effect is the strongest for muNEP could be due to that the altered binding of muNEP compared to NEP is exactly on the amino acid 22 which is the aa that is mutated in the arctic Aβ.
By recombinantly linking the TfR binding moiety (scFv8D3) to our constructs, we could detect high brain uptakes of the NEP protein drug in tg-ArcSwe mice. Brain uptake was 20 times higher compared to the sNEP protein lacking scFv8D3 (Fig. 5A), which is a similar increase as seen with other scFv8D3 fusions to large proteins [17, 39]. In addition, high concentrations were detected in the hippocampus (Fig. 5B), a brain area where Aβ pathology starts [45], and where strong Aβ immunostaining is detected in young tg-ArcSwe mice (Fig. 5C). We have previously also analysed how much of free iodine that enters the brain to see if degradation of constructs is likely to have an effect on the measured uptake but we see that very little free iodine can enter the brain and are therefore not likely to significantly contribute to the signal [65].
The addition of scFc domains prolonged the blood half-life of the NEP-based recombinant proteins compared to the short half-life of soluble NEP. Nevertheless, significantly lower plasma concentrations, twofold shorter blood half-life and fivefold lower drug exposure (AUC blood) of sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3 were displayed compared to scFc-scFv8D3 (Fig. 6C–F). Binding of NEP present in these designs to Aβ, but also other NEP substrates, in the blood followed by subsequent degradation could be one of the possible reasons of the low concentrations of sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3 detected in the blood. The high liver concentrations of sNEP-scFc-scFv8D3 at the early time points (Fig. 5E) further suggest protein degradation. In addition to the low concentrations in the blood, the brain concentrations of NEP proteins drastically decreased 72 h post-injection (Fig. 6B), opposite to what has been previously reported with other Aβ binders containing scFv8D3 [41], suggesting fast clearance of the NEP proteins from the brain. The same pattern of low NEP protein concentration was also evident in the lung and heart (Fig. 6G), two organs containing several NEP substrates [43, 66, 67].
The ability of the recombinant NEP proteins to degrade Aβ in vivo was investigated using tg-ArcSwe mice treated with a single therapeutic dose of 30 nmol/kg. sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3 significantly decreased aggregated Aβ concentration in plasma but not of monomers (Fig. 8A, B). The significant reduction in aggregated plasma Aβ levels did not significantly affect the levels of Aβ aggregates, except oligomers in the brain (Fig. 7). In the brain, we saw a small but significant reduction of Aβ monomers in the TBS-T fraction and of Aβ oligomers as detected by the A11 antibody that detects oligomers with a beta hairpin in the TBS fraction. This beta hairpin is lost when the aggregates grow and form fibrils. The reduction we see in hairpin containing Aβ goes in line with the possibility that neprilysin is efficient in degrading hairpin containing oligomers as we discussed above. Since no significant reduction in the amount of Aβ42 monomers could be detected in the brain, we assume that it is Aβ40 that is reduced. It is also the main type of Aβ in the ArcSwe mice.
There is also a possibility that the effects seen in the brain are due to the reduction of Aβ in the blood, but since it is mainly a reduction in aggregates in the blood and not monomers, we think that is less likely.
Our findings are in accordance with previous studies using similar proteins consisting of recombinant NEP linked to either an albumin protein [68] or the Fc region of IgG antibodies [69]. NEP proteins used in these studies demonstrated no alteration in Aβ concentration in the brain despite the significant Aβ reduction detected in the periphery. However, none of these studies used a BBB transporter and was based on the sink hypothesis of Aβ equilibrium between the brain and periphery. In our study, we used scFv8D3 as the BBB transporter which facilitated the delivery of recombinant NEP proteins into the brain. In addition, treatment with sNEP-scFc-scFv8D3 was associated with a selective reduction (p value 0.03) in membrane-bound Aβ monomers, but not aggregates. These findings are similar to previous studies demonstrating more efficient degradation of Aβ monomers by NEP compared to oligomers [33].
The in vitro experiments demonstrated that NEP-based proteins were capable of degrading arctic-Aβ, opposite to what has been previously reported [70]. Based on the in vitro results, we chose the tg-ArcSwe AD mouse model for the in vivo experiments. Nevertheless, treatment with scFv8D3-fused NEP proteins failed to display any effects on the concentration of arctic-Aβ in tg-ArcSwe mice, but this can be attributed to the ELISA that is set up. With the ELISA used, one will only detect aggregates that have both the N-terminal and the middle available for binding, and likely mostly aggregates will be detected due to the coating with the 3D6 antibody that binds both aggregates and monomers, but that likely will favour aggregates due to the avidity effect. The short retention time of the injected NEP in the brain will likely reduce the possible effect the NEP have had in the brain.
Limitations
One limitation can be the small sample size, especially in a treatment study where n = 3–4 per treatment group. However, since we see this study as a proof of concept demonstrating the treatment effects of BBB-penetrating formats of NEP, we followed the 3R principles and used the minimal number of animals needed to display a statistically significant effect.
Conclusion
Overall, the present study shows the potential of using TfR-mediated transcytosis to successfully deliver NEP proteins into the brain. By adding a scFc and a scFv8D3, our new NEP-based protein designs displayed longer blood half-life and 20 times higher brain uptake compared to sNEP. The retained enzymatic activity in vitro and the significant reduction of Aβ monomers and Aβ oligomers in vivo following a single intravenous injection highlight the potential of recombinant NEP-based proteins in AD although a modified construct with better brain retention would be desired.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Aβ:
-
Amyloid-beta
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- Arc:
-
Arctic APP mutation
- BBB:
-
Blood–brain barrier
- muNEP:
-
Mutated variant of neprilysin (G399V/G714K)
- NEP:
-
Neprilysin
- scFc:
-
Single-chain fragment constant
- scFv:
-
Single-chain fragment variable
- sNEP:
-
Soluble neprilysin
- Swe:
-
Swedish APP mutation
- TfR:
-
Transferrin receptor
- WT:
-
Wild-type
References
Selkoe DJ. Alzheimer’s disease results from the cerebral accumulation and cytotoxicity of amyloid ß-protein. J Alzheimers Dis. 2001;3(1):75–80.
Dawkins E, Small DH. Insights into the physiological function of the β-amyloid precursor protein: beyond Alzheimer’s disease. J Neurochem. 2014;129(5):756–69.
Giuffrida ML, Caraci F, Pignataro B, Cataldo S, Bona PD, Bruno V, et al. β-Amyloid monomers are neuroprotective. J Neurosci. 2009;29(34):10582–7.
Giuffrida ML, Tomasello MF, Pandini G, Caraci F, Battaglia G, Busceti C, et al. Monomeric ß-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front Cell Neurosci. 2015;9:297.
Grimm MOW, Grimm HS, Hartmann T. Amyloid beta as a regulator of lipid homeostasis. Trends Mol Med. 2007;13(8):337–44.
Zimbone S, Monaco I, Gianì F, Pandini G, Copani AG, Giuffrida ML, et al. Amyloid Beta monomers regulate cyclic adenosine monophosphate response element binding protein functions by activating type-1 insulin-like growth factor receptors in neuronal cells. Aging Cell. 2018;17(1):e12684.
Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5.
Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6.
De S, Wirthensohn DC, Flagmeier P, Hughes C, Aprile FA, Ruggeri FS, et al. Different soluble aggregates of Aβ42 can give rise to cellular toxicity through different mechanisms. Nat Commun. 2019;10(1):1541.
Esparza TJ, Wildburger NC, Jiang H, Gangolli M, Cairns NJ, Bateman RJ, et al. Soluble amyloid-beta aggregates from human Alzheimer’s disease brains. Sci Rep. 2016;5(6):38187.
He Y, Zheng MM, Ma Y, Han XJ, Ma XQ, Qu CQ, et al. Soluble oligomers and fibrillar species of amyloid β-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem Biophys Res Commun. 2012;429(3–4):125–30.
Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, et al. Large aggregates are the major soluble Aβ species in AD brain fractionated with density gradient ultracentrifugation. PLoS One. 2012;7(2):e32014.
Sideris DI, Danial JSH, Emin D, Ruggeri FS, **a Z, Zhang YP, et al. Soluble amyloid beta-containing aggregates are present throughout the brain at early stages of Alzheimer’s disease. Brain Commun. 2021;3(3):fcab147.
Spencer B, Rockenstein E, Crews L, Marr R, Masliah E. Novel strategies for Alzheimer’s disease treatment. Expert Opin Biol Ther. 2007;7(12):1853–67.
Tolar M, Abushakra S, Hey JA, Porsteinsson A, Sabbagh M. Aducanumab, gantenerumab, BAN2401, and ALZ-801—the first wave of amyloid-targeting drugs for Alzheimer’s disease with potential for near term approval. Alzheimers Res Ther. 2020;12(1):95.
Campos CR, Kemble AM, Niewoehner J, Freskgård PO, Urich E. Brain shuttle neprilysin reduces central amyloid-β levels. PLoS One. 2020;15(3):e0229850.
Rofo F, Ugur Yilmaz C, Metzendorf N, Gustavsson T, Beretta C, Erlandsson A, et al. Enhanced neprilysin-mediated degradation of hippocampal Aβ42 with a somatostatin peptide that enters the brain. Theranostics. 2021;11(2):789–804.
Sikanyika NL, Parkington HC, Smith AI, Kuruppu S. Powering amyloid beta degrading enzymes: a possible therapy for Alzheimer’s disease. Neurochem Res. 2019;44(6):1289–96.
Miners JS, Barua N, Kehoe PG, Gill S, Love S. Aβ-degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70(11):944–59.
Devault A, Lazure C, Nault C, Le Moual H, Seidah NG, Chrétien M, et al. Amino acid sequence of rabbit kidney neutral endopeptidase 24.11 (enkephalinase) deduced from a complementary DNA. EMBO J. 1987;6(5):1317–22.
Kerr MA, Kenny AJ. The purification and specificity of a neutral endopeptidase from rabbit kidney brush border. Biochem J. 1974;137(3):477–88.
Iwata N, Sekiguchi M, Hattori Y, Takahashi A, Asai M, Ji B, et al. Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Sci Rep. 2013;3(1):1472.
Yasojima K, Akiyama H, McGeer EG, McGeer PL. Reduced neprilysin in high plaque areas of Alzheimer brain: a possible relationship to deficient degradation of β-amyloid peptide. Neurosci Lett. 2001;297(2):97–100.
Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. Expression and functional profiling of neprilysin, insulin degrading enzyme and endothelin converting enzyme in prospectively studied elderly and Alzheimer’s brain. J Neurochem. 2010;115(1):47–57.
Kuruppu S, Rajapakse NW, Minond D, Smith AI. Production of soluble Neprilysin by endothelial cells. Biochem Biophys Res Commun. 2014;446(2):423–7.
Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze beta-amyloid(1–40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides. 1995;16(4):647–52.
Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292(5521):1550–2.
Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of Aβ-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. J Neurosci Res. 2002;70(3):493–500.
Takaki Y, Iwata N, Tsubuki S, Taniguchi S, Toyoshima S, Lu B, et al. Biochemical identification of the neutral endopeptidase family member responsible for the catabolism of amyloid beta peptide in the brain. J Biochem (Tokyo). 2000;128(6):897–902.
Madani R, Poirier R, Wolfer DP, Welzl H, Groscurth P, Lipp HP, et al. Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J Neurosci Res. 2006;84(8):1871–8.
Mouri A, Zou LB, Iwata N, Saido TC, Wang D, Wang MW, et al. Inhibition of neprilysin by thiorphan (i.c.v.) causes an accumulation of amyloid β and impairment of learning and memory. Behav Brain Res. 2006;168(1):83–91.
Newell AJ, Sue LI, Scott S, Rauschkolb PK, Walker DG, Potter PE, et al. Thiorphan-induced neprilysin inhibition raises amyloid beta levels in rabbit cortex and cerebrospinal fluid. Neurosci Lett. 2003;350(3):178–80.
Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, et al. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron. 2003;40(6):1087–93.
Marr RA, Rockenstein E, Mukherjee A, Kindy MS, Hersh LB, Gage FH, et al. Neprilysin gene transfer reduces human amyloid pathology in transgenic mice. J Neurosci Off J Soc Neurosci. 2003;23(6):1992–6.
Oh JH, Choi S, Shin J, Park JS. Protective effect of recombinant soluble neprilysin against β-amyloid induced neurotoxicity. Biochem Biophys Res Commun. 2016;477(4):614–9.
Park MH, Lee JK, Choi S, Ahn J, ** HK, Park JS, et al. Recombinant soluble neprilysin reduces amyloid-beta accumulation and improves memory impairment in Alzheimer’s disease mice. Brain Res. 2013;5(1529):113–24.
Fang XT, Hultqvist G, Meier SR, Antoni G, Sehlin D, Syvänen S. High detection sensitivity with antibody-based PET radioligand for amyloid beta in brain. Neuroimage. 2019;01(184):881–8.
Hultqvist G, Syvänen S, Fang XT, Lannfelt L, Sehlin D. Bivalent brain shuttle increases antibody uptake by monovalent binding to the transferrin receptor. Theranostics. 2017;7(2):308–18.
Syvänen S, Hultqvist G, Gustavsson T, Gumucio A, Laudon H, Söderberg L, et al. Efficient clearance of Aβ protofibrils in AβPP-transgenic mice treated with a brain-penetrating bifunctional antibody. Alzheimers Res Ther. 2018;10(1):49.
Gustafsson S, Gustavsson T, Roshanbin S, Hultqvist G, Hammarlund-Udenaes M, Sehlin D, et al. Blood-brain barrier integrity in a mouse model of Alzheimer’s disease with or without acute 3D6 immunotherapy. Neuropharmacology. 2018;1(143):1–9.
Gustavsson T, Syvänen S, O’Callaghan P, Sehlin D. SPECT imaging of distribution and retention of a brain-penetrating bispecific amyloid-β antibody in a mouse model of Alzheimer’s disease. Transl Neurodegener. 2020;9(1):37.
Fang XT, Hultqvist G, Meier SR, Antoni G, Sehlin D, Syvänen S. High detection sensitivity with antibody-based PET radioligand for amyloid beta in brain. Neuroimage. 2019;1(184):881–8.
D’Elia E, Iacovoni A, Vaduganathan M, Lorini FL, Perlini S, Senni M. Neprilysin inhibition in heart failure: mechanisms and substrates beyond modulating natriuretic peptides. Eur J Heart Fail. 2017;19(6):710–7.
Webster CI, Burrell M, Olsson LL, Fowler SB, Digby S, Sandercock A, et al. Engineering neprilysin activity and specificity to create a novel therapeutic for Alzheimer’s disease. PLoS One. 2014;9(8):e104001.
Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LNG. The Arctic Alzheimer mutation facilitates early intraneuronal Abeta aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27(1):67–77.
Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;4(9):887–93.
Pope D, Madura JD, Cascio M. β-Amyloid and neprilysin computational studies identify critical residues implicated in binding specificity. J Chem Inf Model. 2014;54(4):1157–65.
Fang XT, Sehlin D, Lannfelt L, Syvänen S, Hultqvist G. Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells. Biol Proced Online. 2017;19:11.
Rofo F, Buijs J, Falk R, Honek K, Lannfelt L, Lilja AM, et al. Novel multivalent design of a monoclonal antibody improves binding strength to soluble aggregates of amyloid beta. Transl Neurodegener. 2021;10(1):38.
Englund H, Sehlin D, Johansson AS, Nilsson LNG, Gellerfors P, Paulie S, et al. Sensitive ELISA detection of amyloid-beta protofibrils in biological samples. J Neurochem. 2007;103(1):334–45.
Greenwood F, Hunter W, Glover J. The preparation of 131 I-labelled human growth hormone of high specific radioactivity. Biochem J. 1963;89(1):114–23.
Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–5.
Spencer B, Marr RA, Gindi R, Potkar R, Michael S, Adame A, et al. Peripheral delivery of a CNS targeted, metalo-protease reduces aβ toxicity in a mouse model of Alzheimer’s disease. PLoS One. 2011;6(1):e16575.
Lord A, Englund H, Söderberg L, Tucker S, Clausen F, Hillered L, et al. Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer’s disease transgenic mice. FEBS J. 2009;276(4):995–1006.
Faresjö R, Bonvicini G, Fang XT, Aguilar X, Sehlin D, Syvänen S. Brain pharmacokinetics of two BBB penetrating bispecific antibodies of different size. Fluids Barriers CNS. 2021;18(1):26.
Morrison J, Metzendorf N, Rofo F, Petrovic A, Hultqvist G. A single chain fragment constant (scFc) design enables easy production of a monovalent BBB transporter and provides an improved brain uptake at elevated doses. Submitted. 2022.
** M, O’Nuallain B, Hong W, Boyd J, Lagomarsino VN, O’Malley TT, et al. An in vitro paradigm to assess potential anti-Aβ antibodies for Alzheimer’s disease. Nat Commun. 2018;9(1):2676.
Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350(2):113–6.
Crespi GAN, Hermans SJ, Parker MW, Miles LA. Molecular basis for mid-region amyloid-β capture by leading Alzheimer’s disease immunotherapies. Sci Rep. 2015;5(1):9649.
Hafez D, Huang JY, Huynh AM, Valtierra S, Rockenstein E, Bruno AM, et al. Neprilysin-2 is an important β-amyloid degrading enzyme. Am J Pathol. 2011;178(1):306–12.
Rofo F, Sandbaumhüter FA, Chourlia A, Metzendorf NG, Morrison JI, Syvänen S, et al. Wide-ranging effects on the brain proteome in a transgenic mouse model of Alzheimer’s disease following treatment with a brain-targeting somatostatin peptide. ACS Chem Neurosci. 2021;12(13):2529–41.
Sato K, Tanabe C, Yonemura Y, Watahiki H, Zhao Y, Yagishita S, et al. Localization of mature neprilysin in lipid rafts. J Neurosci Res. 2012;90(4):870–7.
Leissring MA, Lu A, Condron MM, Teplow DB, Stein RL, Farris W, et al. Kinetics of amyloid β-protein degradation determined by novel fluorescence- and fluorescence polarization-based assays *. J Biol Chem. 2003;278(39):37314–20.
Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, et al. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases *. J Biol Chem. 2001;276(24):21895–901.
Rosa A de la, Metzendorf NG, Morrison JI, Faresjö R, Rofo F, Petrovic A, et al. Introducing or removing heparan sulfate binding sites does not alter brain uptake of the blood-brain barrier shuttle scFv8D3. In Review; 2022 Oct [cited 25 Oct 2022]. Available from: https://www.researchsquare.com/article/rs-2166577/v1
Dempsey EC, Wick MJ, Karoor V, Barr EJ, Tallman DW, Wehling CA, et al. Neprilysin null mice develop exaggerated pulmonary vascular remodeling in response to chronic hypoxia. Am J Pathol. 2009;174(3):782–96.
van der Velden VH, Hulsmann AR. Peptidases: structure, function and modulation of peptide-mediated effects in the human lung. Clin Exp Allergy J Br Soc Allergy Clin Immunol. 1999;29(4):445–56.
Henderson SJ, Andersson C, Narwal R, Janson J, Goldschmidt TJ, Appelkvist P, et al. Sustained peripheral depletion of amyloid-β with a novel form of neprilysin does not affect central levels of amyloid-β. Brain. 2014;137(2):553–64.
Walker JR, Pacoma R, Watson J, Ou W, Alves J, Mason DE, et al. Enhanced proteolytic clearance of plasma Aβ by peripherally administered neprilysin does not result in reduced levels of brain Aβ in mice. J Neurosci. 2013;33(6):2457–64.
Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet Lond Engl. 2003;361(9373):1957–8.
Acknowledgements
Biorender.com was used to create some figures.
Radiochemistry and animal work in this study was performed at the SciLifeLab Pilot Facility for Preclinical PET-MRI, a Swedish nationally available imaging platform at Uppsala University, Sweden, financed by the Knut and Alice Wallenberg Foundation.
Funding
Open access funding provided by Uppsala University. This work was supported by grants from Swedish Research Council, Hedlunds stiftelse, Åhlén-stiftelsen, Jeanssons stiftelser, Magnus Bergvallsstiftelse, Vinnova, Alzheimerfonden, Bertil och Ebon Norlins stiftelse, Gunvor och Josef Aners stiftelse, Torsten Söderbergs stiftelse, Konung Gustaf V:s och Drottning Victorias Frimuarestiftelse, Hjärnfonden and Gun and Bertil Stohne’s Foundation.
Author information
Authors and Affiliations
Contributions
FR and GH designed the project. FR and GH designed the protein constructs. FR, NGM, CS and LS produced the antibodies. FR, NGM, CS, LS and AG performed the in vitro assays. FR, NGM, SS and DS performed in vivo work. FR and GH analysed the results and wrote the manuscript with valuable inputs from all the co-authors. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures described in this paper were approved by the Uppsala County Animal Ethical Committee following the ethical guidelines and having the ethical permission number: #5.8.18–13350/17 and #5.8.18–20401/2020.
Consent for publication
Not applicable.
Competing interests
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplementary Figure 1. The sequences of the genes that was inserted in to the pcDNA3.4 vectorused to produce the constructs. The signal peptide is cleaved while expressingthe protein so is not left in the final protein that have been used. A. sNEP-scFc-scFv8D3.B. muNEP-scFc-scFv8D3. C. scFc-scFv8D3. Supplementary Figure 2. Instant thin layer chromatography (iTLC) of proteinsbefore and after in vivo experiment. A. Proteins after labelling with 125I and before application in vivo study. B. Plasmasamples and C. Urine samples 72 hours post injection of 30 nmol/kg body weightof SNEP-scFcscFv8D3, muNEP-scFc-scFv8D3 and scFc-scFv8D, which was appliedintravenously in the tail vein. All samples were applied on a silica-coatedaluminium plate and separated with 70% (v/v) acetone. The radioactive signalwas developed with an X-ray film and red in a Cyclon Phosphoimager. Supplementary Figure 3.Binding selectivity of anti-Ab antibodies used in this study. ELISA plates coatedwith an anti-Ab42capture antibody. Serial dilution of either wild-type Ab1-42 (wt-Ab)or arctic-Ab1-42added to the plates. A: when using 3D6 as the detection antibody, bothwt-Aband arctic-Abcould be detected. B: when using m266 as the detection antibody, bothwt-Aband arctic-Abcould be detected. C: when using mAb27 as the detection antibody, onlyarctic-Abcould be detected. Supplementary Figure 4. Inhibition ELISA demonstrating the binding strengthof m266 antibody to different species of Ab.Five different Ab species were used: Ab1-40monomers, Ab1-40 dimers, Ab1-42oligomers, Ab1-42 protofibrils and Ab1-42fibrils, prepared as described previously [40]. The assay was performed as described previously [40]. Inhibitory concentration-50 (IC50) of m266 bindingto the different Ab species is present in the table. m266 bound strongerto Ab monomers compared to other Abspecies. Binding strength of m266 antibody decreased as the size of Abspecies increased. Supplementary Figure 5. Total concentration of Ab40 and Ab42 in FA soluble brainextracts of tg-ArcSwe mice following treatment with therapeutic doses of 30nmol/kg body weight of sNEP-scFc-scFv8D3 or muNEP-scFc-scFv8D3, usingscFc-scFv8D3 as the negative control. No significant differences detected amongthe three groups. Results presented as mean ±SD. One-way ANOVA with Bonferroni’s multiple comparison test was applied(n=4/sNEP-scFc-scFv8D3 and muNEP-scFc-scFv8D3; n=3/scFc-scFv8D3).(p>0.05=ns; p≤0.05= *; p≤0.01= **; p≤0.001= ***). Supplementary Figure 6. Complete image of the SDS-PAGE gel presented in Fig.2A.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Rofo, F., Metzendorf, N.G., Saubi, C. et al. Blood–brain barrier penetrating neprilysin degrades monomeric amyloid-beta in a mouse model of Alzheimer’s disease. Alz Res Therapy 14, 180 (2022). https://doi.org/10.1186/s13195-022-01132-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13195-022-01132-2