
Abstract
Background
Restraining or slowing ageing hallmarks at the cellular level have been proposed as a route to increased organismal lifespan and healthspan. Consequently, there is great interest in anti-ageing drug discovery. However, this currently requires laborious and lengthy longevity analysis. Here, we present a novel screening readout for the expedited discovery of compounds that restrain ageing of cell populations in vitro and enable extension of in vivo lifespan.
Methods
Using Illumina methylation arrays, we monitored DNA methylation changes accompanying long-term passaging of adult primary human cells in culture. This enabled us to develop, test, and validate the CellPopAge Clock, an epigenetic clock with underlying algorithm, unique among existing epigenetic clocks for its design to detect anti-ageing compounds in vitro. Additionally, we measured markers of senescence and performed longevity experiments in vivo in Drosophila, to further validate our approach to discover novel anti-ageing compounds. Finally, we bench mark our epigenetic clock with other available epigenetic clocks to consolidate its usefulness and specialisation for primary cells in culture.
Results
We developed a novel epigenetic clock, the CellPopAge Clock, to accurately monitor the age of a population of adult human primary cells. We find that the CellPopAge Clock can detect decelerated passage-based ageing of human primary cells treated with rapamycin or trametinib, well-established longevity drugs. We then utilise the CellPopAge Clock as a screening tool for the identification of compounds which decelerate ageing of cell populations, uncovering novel anti-ageing drugs, torin2 and dactolisib (BEZ-235). We demonstrate that delayed epigenetic ageing in human primary cells treated with anti-ageing compounds is accompanied by a reduction in senescence and ageing biomarkers. Finally, we extend our screening platform in vivo by taking advantage of a specially formulated holidic medium for increased drug bioavailability in Drosophila. We show that the novel anti-ageing drugs, torin2 and dactolisib (BEZ-235), increase longevity in vivo.
Conclusions
Our method expands the scope of CpG methylation profiling to accurately and rapidly detecting anti-ageing potential of drugs using human cells in vitro, and in vivo, providing a novel accelerated discovery platform to test sought after anti-ageing compounds and geroprotectors.
Similar content being viewed by others


Background
Organismal ageing has been attributed to nine causal hallmarks [1] which include dysregulated nutrient sensing, increased senescent cell burden, and epigenetic alterations such as changes in DNA methylation. The latter underpins a plethora of epigenetic clocks derived from selected CpG sites which enable the accurate measurement of human age [2,3,4,5]. Consequently, slowing ageing hallmarks at the cellular level have been proposed as a route to increase organismal lifespan with the potential for increased healthspan. In support of such a strategy, there are multiple examples of genetic interventions that can extend both lifespan and healthspan in model organisms. For example, the downregulation of major cellular nutrient signalling pathways, such as glucose-sensing insulin signalling or amino acid-sensing target-of-rapamycin signalling, results in longevity and health improvement in all model organisms tested from yeast to mammals [1]. Furthermore, the selective clearance of senescent cells also extends lifespan in mice [6] and has been shown to alleviate features of an ever-growing number of age-related diseases [7, 8]. Taken together, these seminal findings show that ageing is a malleable process which has the potential to be influenced via pharmacological interventions.
Despite this promise, to date, there are only a handful of pharmacological treatments which have reliably extended lifespan in mammals. One well-studied example is rapamycin, which delays ageing in yeast, Drosophila, C. elegans, and mice [9,10,11,12,13]. Rapamycin has also been shown to slow age-related diseases in multiple species including humans [14]. Interestingly, there is emerging evidence that pharmacological interventions which extend lifespan may have the potential to slow epigenetic clocks. Epigenetic clocks are molecular estimators of biological age based on the DNA methylation levels of specific CpG sites across the genome [2]. For example, rapamycin slowed epigenetic ageing of a 148 CpG clock in mice [15]. However, it should be noted that daily rapamycin treatment over 2.5–3 years did not slow epigenetic ageing in marmosets, albeit the effectiveness of rapamycin-mediated lifespan extension in these experiments is yet to be reported [16].
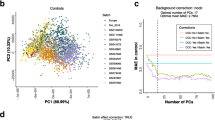
The impact of pharmacological interventions has also been explored at the cellular level. Complementing the in vivo findings above, rapamycin can delay the onset of senescence following serial passaging in human BJ fibroblasts [17] and gingival fibroblasts [The CellPopAge Clock detected decelerated ageing of human primary cells treated with either mTOR, MEK/ERK, or PI3K pathway inhibitors Having built a precise epigenetic clock that measures methylation changes during ageing of adult human primary cells in vitro, we tested if anti-ageing drug treatment of HMFs and HDFs decelerated the CellPopAge Clock. We chose an mTOR inhibitor, rapamycin, which is one of the most robust and evolutionarily conserved anti-ageing drug targets [40], which mediates its effect through downregulation of S6K and Pol III and upregulation of autophagy [9, 41]. We specifically selected a relatively low rapamycin concentration (5 nM). Treatment with this dose did not significantly affect the total number of population doubles observed over 11 weeks of culture when compared to the EtOH solvent control (Additional file 1: Fig. S1A and S1E-G) but did reduce mTOR signalling, as evidenced by decreased pS6K and p4E-BP phosphorylation (Additional file 1: Fig. S4). This setup intentionally mimicked the pro-longevity effects of rapamycin in vivo where it is well accepted that only mild nutrient sensing pathway inhibition increases lifespan and healthspan [1, 42]. DNA methylation profiles from HMFs and HDFs collected following 7, 9, and 11 weeks of 5 nM rapamycin treatment (passages 16, 18 and 20; Fig. 2) were analysed using the CellPopAge Clock and clearly demonstrated that rapamycin slows down methylation changes associated with ageing of cell populations. Interestingly, this clock deceleration was more pronounced upon longer treatment as shown by the gradual decrease of predicted-actual passage from passages 16 to 20. Importantly, when we compare the cumulative population doublings (CPDs) over the extended treatment period with 5 nM rapamycin (Additional file 1: Fig. S1E-G), we see no significant difference compared to the EtOH control (CPDs EtOH: 25.52 ± 1.29; 5 nM rapamycin: 24.12 ± 0.67; p = 0.17, t-test) nor do we see a significant difference in CPDs between rapamycin and EtOH controls if we perform simple linear regression analysis on passage 16 to passage 21 (p = 0.84, Additional file 1: Fig. S1E). Importantly, the difference between rapamycin samples and controls was pronounced when predicted passage and cumulative population doublings are compared (Additional file 1: Fig. S5A-B), with rapamycin samples estimated by the CellPopAge Clock to be four passages younger than the control. We observed a similar pattern for HMFs and HDFs (Fig. 2), suggesting that the CellPopAge Clock is applicable to different in vitro models and for cells isolated from two different tissues (breast and skin), albeit calibration is required for cells that reach senescence at different rates. Using the CellPopAge Clock for the detection of anti-ageing drugs. A Schematic illustrating the experimental set-up conducted in P9 to P20 HDFs and HMFs, passaged weekly and continuously treated with either rapamycin, trametinib, torin2, or dactolisib/BEZ235, represented as a pill. Control cells were treated with vehicle, either DMSO or ethanol. B The CellPopAge Clock predictions of human dermal fibroblasts (HDF) and C human mammary fibroblasts (HMF). Represented is predicted-actual passage for passages 16, 18, and 20, showing deceleration of the CellPopAge Clock upon treatment with anti-ageing drugs rapamycin (5 nM), trametinib (0.1 nM), torin2 (5 nM), and dactolisib/BEZ235 (10 nM) and non-treated control samples (black dots) We then focused on HMFs to test another anti-ageing drug, trametinib [43], an inhibitor of the MEK/ERK signalling pathway, which was shown to extend lifespan in Drosophila [43]. When applied in low concentration, trametinib did not affect cell proliferation and population doublings (Additional file 1: Fig. S1B and S4), and the CellPopAge Clock analysis of trametinib treatment showed clock deceleration for all three passages tested (Fig. 2); therefore, under these conditions, the CellPopAge Clock output was solely affected by ageing of the population in vitro. Next, we sought to test if the CellPopAge Clock has utility for the identification of new compounds which could decelerate ageing of cell populations. We examined the effect of two other inhibitors of nutrient-sensing pathways as mutations in these pathways in model organisms represent the most evolutionary conserved anti-ageing interventions [1]. We tested dactolisib/BEZ235, a dual ATP competitive PI3K and mTOR inhibitor, for which we again optimised the dose of the treatment to obtain a reduction in signalling, as shown by pS6K downstream target 4E-BP (Additional file 1: Fig. S4), without significant proliferation impairment (Additional file 1: Fig. S1C). Dactolisib/BEZ235 slowed down the DNA methylation changes similar to rapamycin, suggesting that dactolisib/BEZ235 could be a new anti-ageing drug according to the output of the CellPopAge Clock (Fig. 2). We also tested torin2, which is a selective ATP-competitive inhibitor of the mTOR pathway that inhibits both mTORC1 and mTORC2, unlike rapamycin, which targets solely mTORC1 [44]. Owing to its more complete inhibition of the mTOR pathway, we were interested in examining its effect on ageing of cell populations, especially as the role of mTORC2 in ageing is less well established. The impact of mTORC2 inhibition on lifespan can be positive or negative depending on which of the mTORC2 downstream effectors is affected, in which tissue, and whether females or male mice are used for the experiment [45]. Some of the negative effects of mTOR pathway inhibition, such as insulin resistance and hyperlipidaemia, are attributed to the mTORC2 branch of the pathway and may arise under certain conditions of prolonged and/or high-dose rapamycin treatment [45]. Interestingly, whilst our CellPopAge Clock suggests that torin2 is indeed a novel anti-ageing drug (Fig. 2 and Additional file 1: Fig. S1D and S4), its effect on the epigenetic ageing of mammalian cell culture appears to be less pronounced than that of rapamycin. This is in line with literature suggesting that a promising strategy to improve healthy ageing is the development of inhibitors that are highly specific for mTORC1 or that target mTORC1 downstream effectors separately [45]. Next, we compared our anti-ageing drug screening results obtained by the CellPopAge Clock with analyses using Horvath’s Multi-Tissue and Skin and Blood clocks, as well as the PhenoAge clock. These clocks did not detect any significant effect of anti-ageing drug treatment (Additional file 1: Fig. S6). The Skin and Blood clock [20] was used recently to measure deceleration of ageing in primary fibroblasts [19, 35]. However, the concentration of rapamycin used in our conditions was five times lower and importantly did not affect cell proliferation, highlighting the sensitivity of our epigenetic clock to detect age-related methylation changes at very low drug concentrations. Under our conditions, the only epigenetic clock that detected gradual methylation changes from passage 10 to passage 20 was the PhenoAge clock (Additional file 1: Fig. S6). However, its output was more variable between samples and inconsistent for anti-ageing drug treatments, reporting both clock acceleration and deceleration. For instance, rapamycin-, dactolisib/BEZ235-, and torin2-treated cells appeared slightly younger compared to controls, whereas trametinib-treated cells were estimated older to some extent (Additional file 1: Fig. S6), unlike the results we obtained with our CellPopAge Clock (Fig. 2). Overall, the CellPopAge Clock that we developed here was more consistent and performed significantly better at determining the age of cells in culture and following known anti-ageing drug treatments compared to existing clocks. Our results are supportive of clocks being highly specialised for a certain task and suggests that whilst other popular epigenetic clocks perform remarkably well for determining donor’s age in years and their health status, they were not able to robustly detect delayed ageing of human primary cells induced by drug treatment over a short period of time in vitro. Next, we assessed if drugs that decelerate the CellPopAge Clock also reduce features associated with senescence, such as morphological changes and expression of senescence and ageing biomarkers [46]. Rapamycin, dactolisib/BEZ235, and trametinib treatment slowed down morphological alteration in cells that gradually occur during ageing of cell populations, namely cell elongation, increased nuclear area and cell area, and the treated cells appearing particularly ‘youthful’ (Fig. 3). Another characteristic of senescence is increased expression of the cyclin-dependent kinase inhibitors p21CIP1/Waf1 and p16INK4a. p21CIP1/Waf1 triggers G1 cycle arrest upon DNA damage and can lead to senescence or apoptosis [8, 47]. Expression of p16INK4a, which is produced from the CDKN2A gene together with p19ARF (p14ARF in humans), increases exponentially during in vivo ageing and has been suggested to stabilise the senescent state [48]. p16INK4a expression was also the marker of choice for senescent cell clearance leading to prolonged lifespan in mice [49]. Treatment with anti-ageing drugs decreases markers of senescence. A Schematic illustrating the experimental set-up conducted in P10 to P22 HMFs, passaged weekly. B Multi-parameter analysis of senescence markers. Robust Z scores were calculated for a panel of measures relative to the vehicle control. Colour coding used to illustrate the number of Z scores of the experimental drug value from the respective vehicle control mean. Scores highlighted in blue denote a shift towards a more proliferative phenotype and scores highlighted in yellow denote a shift to a more senescent phenotype, all with a robust Z scores of ± 0.5. White indicates no change. N = 2 for all except for SA-β-gal. C P22 HMFs stained with DAPI (blue) and Cell Mask, p21, p16, IL-6, or nucleolin (red), or SA-β-Gal (blue) following 96-day treatment with 5 nM rapamycin, 10 nM dactolisib/BEZ235, 0.1 nM trametinib, or their respective controls. Size bar, 100 μm Our results demonstrate that drugs which decelerate the CellPopAge Clock at the same time reduce expression of both nuclear p21CIP1/Waf1 and p16INK4a compared to non-treated cells, showing their efficacy in delaying the senescence programme (Fig. 3B, C and Additional file 5: Table S5). In addition, one of the most frequently used senescent markers, senescence-associated β-galactosidase activity (SA-β-Gal), was decreased upon anti-ageing drugs treatment with rapamycin and dactolisib/BEZ235, but not in cells treated with trametinib (Fig. 3B, C). Another difference in senescent markers was observed with interleukin-6 (IL-6), which is one of the most important inflammatory cytokines and part of the senescence-associated secretory phenotype. IL-6 was significantly reduced in aged cells upon rapamycin and dactolisib/BEZ235 treatment but not in trametinib-treated cells (Fig. 3B, C). This difference possibly stems from the overactivated RAS/ERK pathway being a more prominent inducer of senescence than the overactivated mTOR/PI3K pathway [50], and hence corresponding inhibitors have different potency in inhibiting senescence. Finally, we examined the nucleolus, an organelle dedicated to rRNA production and ribosomal assembly, as it has recently emerged that maintenance of its structure, and low levels of nucleolar methyltransferase fibrillarin, is a common denominator for major anti-ageing intervention from worms to mice [51]. We observed that as a consequence of ageing, nucleoli in aged HMFs lose their defined round shape and are more diffused, with dimmer DAPI staining. For rapamycin and dactolisib/BEZ235, we observed clearly defined and ‘younger’ looking nucleoli in aged cells. However, trametinib-treated cells resembled the nucleoli of controls. In summary, a panel of the most frequently used markers for cell senescence confirmed that drugs which decelerate the CellPopAge Clock also make the cells appear more youthful. This strongly suggests that the CellPopAge Clock can be used as a robust and sensitive detector of compounds which delay ageing of cell populations in vitro. Having discovered two potential novel drug treatments which delay ageing of cells in vitro using the CellPopAge Clock, we then extended our discovery platform by asking if dactolisib/BEZ235 or torin2 could influence ageing at the organismal level. This is important as tissue-specific drug toxicity, which can be missed in cell culture, is one of the major reasons for drug failure in clinical trials. To support an expedited platform, we took advantage of the model organism the fruit fly Drosophila melanogaster, which has well-described characteristics of cellular and organismal ageing [52]. For in vivo longevity studies, we used the outbred wild-type wDah strain which is particularly suitable for ageing studies, Drosoflipper device for fast fly transfer, and specially formulated holidic medium [31] to increase drug bioavailability compared to standard sugar-yeast-agar fly food. We used rapamycin as a positive control for these longevity experiments and showed that median lifespan extension on holidic media varied from 7 to 9% compared to ethanol solvent control, depending on 1 μM or 5 μM concentration, respectively (p < 0.001, log-rank test), which is comparable to published literature [53] (Fig. 4A). Importantly, both dactolisib/BEZ235 and torin2 significantly extended lifespan in Drosophila by 7% (p < 0.001, log-rank test) (Fig. 4B, C). This firmly demonstrates that drugs that decelerate the CellPopAge Clock have similarly favourable output on major anti-ageing biomarkers in vitro and extend longevity in vivo (Fig. 4D). Drugs that decelerate the CellPopAge Clock extend lifespan in vivo. A Lifespan analysis on wDah background wild-type flies fed with holidic food containing different concentration of rapamycin or ethanol as solvent control. For each condition, 150 flies were used. B Lifespan analysis on wDah background wild-type flies fed with holidic food containing different concentration of dactolisib/BEZ235 or DMSO as solvent control. For each condition, 150 flies were used. C Lifespan analysis on wDah background wild-type flies fed with holidic food containing different concentration of torin2 or DMSO as solvent control. For each condition, 150 flies were used. D Schematic representation of our proposed screening platform which combines our novel CellPopAge Clock and other ageing biomarkers in vitro primary human cell, together with in vivo Drosophila lifespan experiments, for a detailed and robust capture of anti-ageing drug potential
The CellPopAge Clock as a tool for the identification of compounds which decelerates ageing of cell populations
Delayed ageing in human primary cells treated with anti-ageing compounds, measured by the CellPopAge Clock, is accompanied by reduction of senescence and ageing biomarkers

The two newly discovered anti-ageing compounds, torin2 and dactolisib/BEZ235, extend lifespan in vivo in Drosophila

Discussion
We present the CellPopAge Clock as the first robust epigenetic clock for the rapid discovery of anti-ageing drugs directly in human cells in vitro. We focused on the discovery of anti-ageing compounds which decelerated epigenetic ageing of cell populations in vitro at very low doses but which did not influence cellular proliferation. Moreover, we utilised relatively low-dose treatments of established and novel anti-ageing compounds to minimise potential off-target effects. Another relevant aspect of our design was to perform these lengthy experiments in the absence of antibiotics. Next, we coupled these anti-ageing compounds with in vivo Drosophila lifespan experiments as this model organism significantly shortens the discovery time compared to 3-year long mice longevity analysis. The rationale for this approach was to identify compounds with in vivo utility which uncoupled epigenetic ageing from cellular division.
Taken together, the CellPopAge Clock, based on 42 CpG sites, is a trained, tested, and validated epigenetic clock with underlying algorithm, which we believe is unique among existing epigenetic clocks as it is designed to detect compounds that restrain ageing of cell populations in vitro. Coupled with in vivo lifespan experiments in Drosophila, our pipeline represents a novel screening platform for the expedited discovery of anti-ageing compounds (Fig. 4D).
Initially, we benchmarked our CellPopAge Clock against other available epigenetic clocks: the Multi-Tissue clock, the PhenoAge clock, and the Skin and Blood clock. The latter was developed to measure cellular age in vitro. However, none of these epigenetic age clocks could accurately detect the effect of low-dose, short-term anti-ageing drugs on cells in vitro. Thus, our CellPopAge Clock differs from these previously published clocks in being able to measure interventions which slow both epigenetic ageing of cell populations and the accumulation of biomarkers of senescence and age across a range of different cell types. This is encouraging considering the breadth of evidence that associates cellular ageing, or senescence, with ageing and diseases of advanced age.
To provide in vivo utility for the CellPopAge Clock as a readout, we took advantage of Drosophila to assess the potential effect of novel anti-ageing compounds on lifespan. We selected this model as lifespan studies are comparatively short at ~ 90 days, thereby providing an expedited and relatively inexpensive in vivo model for testing potentially novel anti-ageing compounds. The development of formulated holidic medium was considered a further advantage as it provided enhanced drug bioavailability. These studies enabled us to utilise the well-established lifespan extender, rapamycin, as a benchmark, and to validate dactolisib/BEZ235 and torin2 as novel anti-ageing compounds. It should be noted that the CellPopAge Clock was not designed to measure intrinsic cell ageing or the biological age of cells in vivo.
Taken together, our results show that by using the CellPopAge Clock, cultured primary human cells can be used to measure ageing of these populations and can reliably detect anti-ageing effects upon a relatively brief, low-dose treatment. By doing so, this fast and accurate screening platform is expected to accelerate the discovery of novel preventive treatments for age-related disease, directly using human cells in vitro and in vivo lifespan experiments using Drosophila. Future refinements could include the use of multiplex DNA methylation-based PCR for the 42 CpG sites of the CellPopAge Clock will facilitate a screening assay for expedited drug discovery. Importantly, follow-up research should be focused on expanding our findings to different types of primary cells from donors of different ages as well as on testing further compounds. Whilst ageing itself is not a disease, potential anti-ageing drugs could be FDA approved separately for different conditions. For example, the first study to test broad-spectrum protection capacity of metformin, the TAME study, is underway [54]. In addition, it was shown that rapamycin/Everolimus pre-treatment dramatically improves flu vaccination and immune response in the elderly [55]. In mice, it also lowers the incidence of tumours [56], and it shows promising results in the field of neurodegeneration [57]. This supports the idea that targeting healthy ageing at the cellular level might have multiple beneficial outputs.
Given the wealth of information stored in our epigenome, we expect a range of biological outputs to be extracted by the CellPopAge Clock and other epigenetic clock algorithms in the future. For example, testing different compounds using the CellPopAge Clock could potentially reveal new anti-ageing pathways, currently dominated by IIS and mTOR signalling pathways. Such approaches could also help us to improve our knowledge base of not only ageing biology but of the molecular pathways underpinning the epigenetic clocks, understanding of which is limited. Our experimental setup is also suitable for nutraceutical approaches whereby dietary supplements can be rigorously tested for their effect on ageing.
At present, particularly interesting findings are emerging from the dissection of the different modules that epigenetic clocks are made of [4, 58], as well as from single cell epigenetic clock analysis [59], both of which will contribute to our mechanistic understanding of epigenetic clocks. Thus, it is interesting to speculate that a range of individual cellular hallmarks of ageing, including development and maintenance programmes, circadian rhythms, metabolism, and DNA damage and repair, might all interdependently cause gradual deterioration of CpG methylation with ageing. Focused epigenetic clock algorithms [4] through the use of in vitro systems offer a route to identifying these individual cellular fingerprints and to exploring this hypothesis. In summary, we expect our novel discovery platform to accelerate therapeutic innovation for strongly sought-after anti-ageing drugs and geroprotective strategies to improve healthy human ageing.
Conclusions
In the present study, we have developed a novel DNA methylation clock, the CellPopAge Clock, specialised for determining age in population of primary cells in culture, for the purpose of drug screening and in the long-term develo** treatments that can improve health during ageing.
Availability of data and materials
All methylation microarray data reported in this study have been deposited in the ArrayExpress (https://www.ebi.ac.uk/arrayexpress/) public repository and are accessible under accession number E-MTAB-8327 (https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-8327?query=E-MTAB-8327) [23]. The CellPopAge Clock is available from GitHub at https://github.com/ucl-medical-genomics/CellPopAge-epigenetic-clock [30]. Correspondence and requests for materials should be addressed to IB, CLB, RL, and SB.
Abbreviations
- 3′UTR:
-
Three prime untranslated region
- 4E-BP:
-
Eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1
- CDKN2A:
-
Cyclin-dependent kinase inhibitor 2A
- CPD:
-
Cumulative population doubling
- DAPI:
-
4′,6-Diamidino-2-phenylindole
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- DMSO:
-
Dimethyl sulfoxide
- DTT:
-
DL-dithiothreitol
- EDTA:
-
Ethylenediaminetetraacetic acid
- EtOH:
-
Ethanol
- FBS:
-
Foetal bovine serum
- FDA:
-
Food and Drug Administration
- GGN:
-
Gametogenetin
- GRID1:
-
Glutamate ionotropic receptor delta type subunit 1
- HDFs:
-
Human dermal fibroblasts
- HMFs:
-
Human mammary fibroblasts
- hTERT:
-
Human telomerase reverse transcriptase
- IGRs:
-
Intergenic regions
- IIS:
-
Insulin/IGF-1 signalling pathway
- IL-6:
-
Interleukin-6
- LDLRAD4:
-
Low-density lipoprotein receptor class A domain containing 4
- MEIS2:
-
Meis homeobox 2
- MEK/ERK:
-
Mitogen-activated protein kinase/extracellular signal-regulated kinase
- MSCs:
-
Mesenchymal stem cells
- MTA:
-
Material transfer agreement
- mTOR:
-
Mammalian or mechanistic target of rapamycin
- NF1:
-
Neurofibromin 1
- NPSR1:
-
Neuropeptide S receptor 1
- PCR:
-
Polymerase chain reaction
- PI3K:
-
Phosphoinositide 3-kinases
- Pol III:
-
RNA polymerase III
- PROP1:
-
PROP paired-like homeobox 1
- RFX4:
-
Regulatory factor X4
- RMSE:
-
Root mean square error
- RUNX3:
-
RUNX family transcription factor 3
- S6K:
-
Ribosomal protein S6 kinase
- SA-β-Gal:
-
Senescence-associated β-galactosidase activity
- SMARCA2:
-
SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A, member 2
- SYA:
-
Sugar/yeast/agar
- TAME:
-
Targeting Aging with Metformin
- TOR:
-
Target of rapamycin
References
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217.
Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19:371–84.
Raj K, Horvath S. Current perspectives on the cellular and molecular features of epigenetic ageing. Exp Biol Med (Maywood). 2020;245:1535370220918329.
Seale K, Horvath S, Teschendorff A, Eynon N, Voisin S. Making sense of the ageing methylome. Nat Rev Genet. 2022;23:585–605.
Bell CG, Lowe R, Adams PD, Baccarelli AA, Beck S, Bell JT, Christensen BC, Gladyshev VN, Heijmans BT, Horvath S, et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 2019;20:249.
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479:232–6.
Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96.
He S, Sharpless NE. Senescence in health and disease. Cell. 2017;169:1000–11.
Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab. 2010;11:35–46.
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–5.
Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261.
Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–84.
Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK. TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO. Cell Metab. 2012;15:713–24.
Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. 2014;6:268ra179.
Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 2017;18:57.
Horvath S, Zoller JA, Haghani A, Lu AT, Raj K, Jasinska AJ, Mattison JA, Salmon AB. DNA methylation age analysis of rapamycin in common marmosets. Geroscience. 2021;43:2413–25.
Kolesnichenko M, Hong L, Liao R, Vogt PK, Sun P. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11:2391–401.
**a Y, Sun M, **e Y, Shu R. mTOR inhibition rejuvenates the aging gingival fibroblasts through alleviating oxidative stress. Oxid Med Cell Longev. 2017;2017:6292630.
Horvath S, Lu AT, Cohen H, Raj K. Rapamycin retards epigenetic ageing of keratinocytes independently of its effects on replicative senescence, proliferation and differentiation. Aging (Albany NY). 2019;11:3238–49.
Horvath S, Oshima J, Martin GM, Lu AT, Quach A, Cohen H, Felton S, Matsuyama M, Lowe D, Kabacik S, et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford progeria syndrome and ex vivo studies. Aging (Albany NY). 2018;10:1758–75.
Koch CM, Joussen S, Schellenberg A, Lin Q, Zenke M, Wagner W. Monitoring of cellular senescence by DNA-methylation at specific CpG sites. Aging Cell. 2012;11:366–9.
Lowe R, Overhoff MG, Ramagopalan SV, Garbe JC, Koh J, Stampfer MR, Beach DH, Rakyan VK, Bishop CL. The senescent methylome and its relationship with cancer, ageing and germline genetic variation in humans. Genome Biol. 2015;16:194.
Lowe R, Ecker S: A CellPopAge epigenetic clock for expedited discovery of anti-ageing compounds in vitro. ArrayExpress 2024:https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-8327?query=E-MTAB-8327.
Fortin JP, Triche TJ Jr, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics. 2017;33:558–60.
McCartney DL, Walker RM, Morris SW, McIntosh AM, Porteous DJ, Evans KL. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom Data. 2016;9:22–4.
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115.
Levine ME, Lu AT, Quach A, Chen BH, Assimes TL, Bandinelli S, Hou L, Baccarelli AA, Stewart JD, Li Y, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY). 2018;10:573–91.
Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1–22.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Lowe R, Ecker S: CellPopAge-epigenetic-clock. GitHub 2024:https://github.com/ucl-medical-genomics/CellPopAge-epigenetic-clock.
Piper MD, Blanc E, Leitao-Goncalves R, Yang M, He X, Linford NJ, Hoddinott MP, Hopfen C, Soultoukis GA, Niemeyer C, et al. A holidic medium for Drosophila melanogaster. Nat Methods. 2014;11:100–5.
Tyler EJ, Gutierrez Del Arroyo A, Hughes BK, Wallis R, Garbe JC, Stampfer MR, Koh J, Lowe R, Philpott MP, Bishop CL. Early growth response 2 (EGR2) is a novel regulator of the senescence programme. Aging Cell. 2021;20:e13318.
Avelar RA, Ortega JG, Tacutu R, Tyler EJ, Bennett D, Binetti P, Budovsky A, Chatsirisupachai K, Johnson E, Murray A, et al. A multidimensional systems biology analysis of cellular senescence in aging and disease. Genome Biol. 2020;21:91.
Wallis R, Milligan D, Hughes B, Mizen H, Lopez-Dominguez JA, Eduputa U, Tyler EJ, Serrano M, Bishop CL. Senescence-associated morphological profiles (SAMPs): an image-based phenotypic profiling method for evaluating the inter and intra model heterogeneity of senescence. Aging (Albany NY). 2022;14:4220–46.
Sturm G, Cardenas A, Bind MA, Horvath S, Wang S, Wang Y, Hagg S, Hirano M, Picard M. Human aging DNA methylation signatures are conserved but accelerated in cultured fibroblasts. Epigenetics. 2019;14:961–76.
Field AE, Robertson NA, Wang T, Havas A, Ideker T, Adams PD. DNA methylation clocks in aging: categories, causes, and consequences. Mol Cell. 2018;71:882–95.
Yang Z, Wong A, Kuh D, Paul DS, Rakyan VK, Leslie RD, Zheng SC, Widschwendter M, Beck S, Teschendorff AE. Correlation of an epigenetic mitotic clock with cancer risk. Genome Biol. 2016;17:205.
Teschendorff AE. A comparison of epigenetic mitotic-like clocks for cancer risk prediction. Genome Med. 2020;12:56.
Endicott JL, Nolte PA, Shen H, Laird PW. Cell division drives DNA methylation loss in late-replicating domains in primary human cells. Nat Commun. 2022;13:6659.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–76.
Filer D, Thompson MA, Takhaveev V, Dobson AJ, Kotronaki I, Green JWM, Heinemann M, Tullet JMA, Alic N. RNA polymerase III limits longevity downstream of TORC1. Nature. 2017;552:263–7.
Bjedov I, Partridge L. A longer and healthier life with TOR down-regulation: genetics and drugs. Biochem Soc Trans. 2011;39:460–5.
Slack C, Alic N, Foley A, Cabecinha M, Hoddinott MP, Partridge L. The Ras-Erk-ETS-signaling pathway is a drug target for longevity. Cell. 2015;162:72–83.
Liu Q, Xu C, Kirubakaran S, Zhang X, Hur W, Liu Y, Kwiatkowski NP, Wang J, Westover KD, Gao P, et al. Characterization of Torin2, an ATP-competitive inhibitor of mTOR, ATM, and ATR. Cancer Res. 2013;73:2574–86.
Kennedy BK, Lamming DW. The mechanistic target of rapamycin: the grand conducTOR of metabolism and aging. Cell Metab. 2016;23:990–1003.
Hanzelmann S, Beier F, Gusmao EG, Koch CM, Hummel S, Charapitsa I, Joussen S, Benes V, Brummendorf TH, Reid G, et al. Replicative senescence is associated with nuclear reorganization and with DNA methylation at specific transcription factor binding sites. Clin Epigenetics. 2015;7:19.
McHugh D, Gil J. Senescence and aging: causes, consequences, and therapeutic avenues. J Cell Biol. 2018;217:65–77.
Gire V, Dulic V. Senescence from G2 arrest, revisited. Cell Cycle. 2015;14:297–304.
Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–9.
Kennedy AL, Morton JP, Manoharan I, Nelson DM, Jamieson NB, Pawlikowski JS, McBryan T, Doyle B, McKay C, Oien KA, et al. Activation of the PIK3CA/AKT pathway suppresses senescence induced by an activated RAS oncogene to promote tumorigenesis. Mol Cell. 2011;42:36–49.
Tiku V, Antebi A. Nucleolar function in lifespan regulation. Trends Cell Biol. 2018;28:662–72.
Piper MDW, Partridge L. Drosophila as a model for ageing. Biochim Biophys Acta Mol Basis Dis. 2018;1864:2707–17.
Fan X, Liang Q, Lian T, Wu Q, Gaur U, Li D, Yang D, Mao X, ** Z, Li Y, Yang M. Rapamycin preserves gut homeostasis during Drosophila aging. Oncotarget. 2015;6:35274–83.
Barzilai N, Crandall JP, Kritchevsky SB, Espeland MA. Metformin as a tool to target aging. Cell Metab. 2016;23:1060–5.
Mannick JB, Morris M, Hockey HP, Roma G, Beibel M, Kulmatycki K, Watkins M, Shavlakadze T, Zhou W, Quinn D, et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci Transl Med. 2018;10(449).
Anisimov VN, Zabezhinski MA, Popovich IG, Piskunova TS, Semenchenko AV, Tyndyk ML, Yurova MN, Rosenfeld SV, Blagosklonny MV. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle. 2011;10:4230–6.
Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437–52.
Liu Z, Leung D, Thrush K, Zhao W, Ratliff S, Tanaka T, Schmitz LL, Smith JA, Ferrucci L, Levine ME. Underlying features of epigenetic aging clocks in vivo and in vitro. Aging Cell. 2020;19:e13229.
Trapp AKC, Gladyshev VN. Profiling epigenetic age in single cells. Nature Aging. 2021;1:1189–201.
Stampfer MR, Bartholomew JC, Smith HS, Bartley JC. Metabolism of benzo[a]pyrene by human mammary epithelial cells: toxicity and DNA adduct formation. Proc Natl Acad Sci U S A. 1981;78:6251–5.
Acknowledgements
We wholeheartedly thank members of our laboratories for exciting discussions that significantly improved our manuscript.
Funding
IB acknowledges funding from ERC StG 311331, ERC PoC 842174, Royal Society Research Grant, The Bill Lyons foundation. SB acknowledges support from the Wellcome Trust (218274/Z/19/Z). This work was supported in part by the CRUK-UCL Centre Award (C416/A25145) awarded to IB and SB and by the Radiation Research Unit at the Cancer Research UK City of London Centre Award [C7893/A28990] awarded to IB. CLB acknowledges funding for EJT from the MRC (MR/K501372/1) and BBSRC (BB/P002579/1) and DM from the BBSRC (BB/N503629/1). JG and MS are supported by U.S. Department of Energy (under Contract No. DE-AC02-05CH11231) and the Congressionally Directed Medical Research Programs Breast Cancer Research Program Era of Hope Scholar Award (BC141351).
Author information
Authors and Affiliations
Contributions
SB and IB developed initial concept. SB, IB, RL, and CLB finalised concept develo** and designed the experiments. RL, SE, and APW analysed the data. RL developed the CellPopAge Clock. CLB and EJT provided cell culture expertise. CL, EJT, ERS, VEMM, DM, and IB performed all the experiments. SE, SB, IB, RL, and CLB wrote the manuscript. JCG and MRS provided critical reagents. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Normal finite lifespan human dermal fibroblasts (HDFs) were obtained from face lift dermis following a kind donation from a 42-year-old female, anonymous healthy patient, under standard ethical practice, reference LREC No. 09/HO704/69. Normal finite lifespan adult human mammary fibroblasts (HMFs) were obtained from reduction mammoplasty tissue of a 16-year-old individual, donor 48 [60]. The tissue was obtained in 1980 based on a pathology consent form and distributed to us under a current IRB which explicitly allows distribution of materials.
Consent for publication
All authors approved the final manuscript before submission.
Competing interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
13073_2024_1349_MOESM1_ESM.pdf
Additional file 1: Fig. S1. Drug treatment of HMF did not affect cell growth as measured by population doubling. Fig. S2. Develo** the CellPopAge Clock. Fig. S3. The CellPopAge Clock predictions of Human Dermal Fibroblasts (HDF) immortalized with hTERT. Fig. S4. Western blot analysis on HMFs treated with anti-ageing drugs. Fig. S5. Comparison of cumulative population doublings following treatment with 5 nM rapamycin and control cells that were treated with ethanol as a vehicle control. Fig. S6. Predicted age of control samples and samples treated with potential anti-ageing drugs using three existing epigenetic clocks.
13073_2024_1349_MOESM2_ESM.xlsx
Additional file 2: Table S1. List of 42 CpGs, that were selected using the elastic net regression model as predictors of cell passage.
13073_2024_1349_MOESM3_ESM.pdf
Additional file 3: Table S2. Spearman's rank correlation and RMSE of the different clocks' predictions with actual cell passage of 26 samples that were not used to build the clock.
13073_2024_1349_MOESM4_ESM.xlsx
Additional file 4: Table S3. Testing the CellPopAge Clock in a publicly available dataset from Endicott et al. [37].
13073_2024_1349_MOESM5_ESM.xlsx
Additional file 5: Table S4. The CellPopAge Clock predictions of Human Dermal Fibroblasts (HDF) immortalized with hTERT.
13073_2024_1349_MOESM6_ESM.xlsx
Additional file 6: Table S5. Expression levels of p21, p16, SA-β-Gal, IL-6 and nucleolin in treated and non-treated cells.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lujan, C., Tyler, E.J., Ecker, S. et al. An expedited screening platform for the discovery of anti-ageing compounds in vitro and in vivo. Genome Med 16, 85 (2024). https://doi.org/10.1186/s13073-024-01349-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-024-01349-w