
Abstract
Background
Chromatin modifications such as DNA methylation are targeted to transposable elements by small RNAs in a process termed RNA-directed DNA methylation (RdDM). In plants, canonical RdDM functions through RNA polymerase IV to reinforce pre-existing transposable element silencing. Recent investigations have identified a “non-canonical” form of RdDM dependent on RNA polymerase II expression to initiate and re-establish silencing of active transposable elements. This expression-dependent RdDM mechanism functions through RNAi degradation of transposable element mRNAs into small RNAs guided by the RNA-dependent RNA polymerase 6 (RDR6) protein and is therefore referred to as RDR6-RdDM.
Results
We performed whole-genome MethylC-seq in 20 mutants that distinguish RdDM mechanisms when transposable elements are either transcriptionally silent or active. We identified a new mechanism of expression-dependent RdDM, which functions through DICER-LIKE3 (DCL3) but bypasses the requirement of both RNA polymerase IV and RDR6 (termed DCL3-RdDM). We found that RNA polymerase II expression-dependent forms of RdDM function on over 20 % of transcribed transposable elements, including the majority of full-length elements with all of the domains required for autonomous transposition. Lastly, we find that RDR6-RdDM preferentially targets long transposable elements due to the specificity of primary small RNAs to cleave full-length mRNAs.
Conclusions
Expression-dependent forms of RdDM function to critically target DNA methylation to full-length and transcriptionally active transposable elements, suggesting that these pathways are key to suppressing mobilization. This targeting specificity is initiated on the mRNA cleavage-level, yet manifested as chromatin-level silencing that in plants is epigenetically inherited from generation to generation.
Similar content being viewed by others

Background
Transposable elements (TEs) are mobile fragments of DNA that can generate mutations and genome instability. To repress TE activity and new mutations, cells target TEs for epigenetic transcriptional silencing. Small RNAs (sRNAs) are the triggers of epigenetic transcriptional silencing targeted to transposable elements (TEs) and transgenes. sRNAs are known to direct cytosine DNA methylation and histone tail post-translational modifications in both mice and plants, while in organisms that lack cytosine DNA methylation (such as fission yeast, C. elegans, and Drosophila) sRNAs direct only histone tail modifications (reviewed in [1]). The mechanism of small RNA-directed DNA methylation (RdDM) has been extensively investigated in the reference plant Arabidopsis, where a “canonical” form of RdDM has been uncovered (reviewed in [2]). This canonical form of RdDM begins with the transcription of the target locus by the RNA polymerase protein Pol IV, a plant-specific Pol II paralog [3], which generates a non-coding RNA that is immediately converted into double-stranded RNA (dsRNA) via RNA-dependent RNA polymerase 2 (RDR2). The Pol IV/RDR2 derived dsRNA is cleaved by the RNaseIII DICER protein DCL3 into 23–24 nucleotide (nt) small interfering RNAs (siRNAs) and these 24 nt siRNAs are incorporated into either the Argonaute 4 (AGO4) or AGO6 proteins [4]. In the nucleus, the siRNA-loaded AGO4/AGO6 can base pair with a nascent non-coding RNA still attached to its DNA template produced by Pol V, a second plant-specific paralog of Pol II. The Pol V transcript acts as a scaffold for protein assembly, and interaction between AGO4/6 and the Pol V transcript results in the recruitment of the protein DRM2 to methylate the cytosines of the corresponding locus.
Pol IV is recruited to and transcribes regions of the genome that have reduced histone acetylation, undergo CG-context maintenance methylation, and are enriched for H3K9me2 [5, 6], heterochromatic marks that decorate regions of the genome inhibited for mRNA production. Canonical Pol IV-targeted RdDM (Pol IV-RdDM) is known to reinforce DNA methylation at regions of TE heterochromatin adjacent to genes [7, 8]. Several laboratories have recently investigated how DNA methylation is initiated at a region of the genome that is actively producing an mRNA and is not already silenced. These investigations have uncovered various “non-canonical” mechanisms of RdDM, which do not rely on Pol IV, but rather are triggered by Pol II mRNA transcripts [9–13]. Pol II TE mRNAs can undergo degradation via endogenous RNAi into 21–22 nt siRNAs [14, 15]. In Arabidopsis, the TE mRNA is converted into dsRNA via RDR6, and this dsRNA is cleaved into 21–22 nt siRNAs via DCL4 and DCL2, respectively [15]. Originally thought to be only a post-transcriptional mechanism of silencing, several studies have determined that the degradation products of Pol II-derived mRNAs can trigger RdDM [9, 12, 13, 16]. The best characterized of these pathways is RDR6-RdDM, where the RDR6-dependent 21–22 nt siRNAs are incorporated into the AGO6 protein and drive RdDM in a Pol V and DRM2-dependent manner [16].
There are only a few known targets of RDR6-RdDM [12, 16]. This is due to the fact that this pathway acts on Pol II transcriptionally active regions of the genome and over time these regions become transcriptionally silenced and regulated by either Pol IV-RdDM or by the maintenance methylation pathway that is not dependent on small RNAs [7, 17]. Maintenance methylation occurs separately for each cytosine sequence context, with CG methylation propagated by MET1, CHG (where H = A, C or T) by CMT3, and CHH context methylation by CMT2 [17–19]. Like Pol IV, CMT2 and CMT3 are guided to previously silenced loci by the H3K9me2 heterochromatic mark [17, 20]. CHH context maintenance methylation is low compared to CG or CHG [17] and therefore RdDM (which targets all cytosine contexts equally) has traditionally been assayed by investigating the CHH methylation level [21, 22].
Maintenance methylation of TEs is coordinated by Decrease in DNA methylation 1 (DDM1) [23], a swi/snf family chromatin remodeling protein. DDM1 specifically regulates TEs and in ddm1 mutants TEs undergo loss of H3K9me2, CG DNA methylation, and heterochromatin condensation [23, 24]. This results in genome-wide TE transcriptional activation [23] and the triggering of the RNAi mechanism to degrade TE mRNAs into 21–22 nt siRNAs [15, 25]. In ddm1 mutant plants, TE transcriptional silencing cannot be regained and therefore the cell is stuck in a perpetual cycle of attempted re-silencing via RdDM. Re-targeting of TEs for silencing, and in particular CHH hyper-methylation, is a conserved consequence of TE activation via ddm1 mutation in Arabidopsis, maize, and rice [12, 26, 27]. ddm1 mutants display unmatched resolution of the mechanisms the cell uses to re-silence TEs [28, 29]. Investigation of ddm1 mutants lead to the discovery of RDR6-RdDM [12, 16]; however, the genome-wide roles RDR6-RdDM have been a continued question. For example, what are the additional targets and the overall role of RDR6-RdDM, is this the sole non-canonical RdDM mechanism that functions genome-wide, and why are particular TEs targeted to undergo non-canonical forms of RdDM while others are not? To address these questions, we created a genome-wide DNA methylation and small RNA dataset in 20 key RdDM mutants that span both the TE-silent and TE-active contexts, providing insight to the pathways the plant uses to target DNA methylation to specific TEs.
Results
RDR6-RdDM targets many transcriptionally active TEs
The switch from an epigenetically silenced state to transcriptional activation is known to trigger Pol II expression-dependent mechanisms of TE silencing such as RDR6-RdDM on the single-locus level [12]. To examine genome-wide methylation states of both active and inactive TEs, we generated a dataset containing whole-genome MethylC-seq of nine key RdDM mutant genotypes in the wild-type Columbia (wt Col) background as well as the same nine mutant genotypes in the ddm1 mutant background. TE transcription is globally reactivated in the ddm1 mutant (Additional file 1: Figure S1) [23], whereas the RdDM mutants that we investigated generally do not show TE transcriptional reactivation or at least not nearly as severe of a transcriptional reactivation compared to ddm1. For example, even in pol V mutants, which are defective for all RdDM [30], global TE activation is minimal compared to ddm1 (Additional file 1: Figure S1) [19, 22]. Therefore, in this study any genotype without ddm1 is referred to as the TE-silent context and our dataset distinguishes RdDM types in both the TE-silent context and the globally reactivated ddm1 TE-active context.
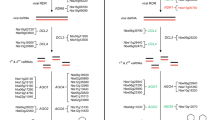
We determined that using only uniquely map** sequencing reads resulted in reduced coverage of repetitive TE regions; however, sequencing coverage was high enough to assay RdDM dynamics of individual TE copies (see “Methods,” Additional file 2: Results, and Additional file 3: Figure S2). To identify the regions of the genome targeted by RDR6-RdDM (and contrast them to the regions regulated by Pol IV-RdDM), we identified differentially methylated regions (DMRs) between all of the genotypes (see “Methods”). Aligning the DMRs, we find that the average wt Col and rdr6 CHH methylation patterns are indistinguishable, demonstrating that RDR6-RdDM plays a minor genome-wide role in the TE-silent context (Fig. 1a, replicate data in Additional file 4: Figure S3A). In contrast, pol IV mutants lose methylation from the DMRs, confirming that Pol IV-RdDM functions to target CHH methylation on a genome-wide level in the TE-silent context (Fig. 1a) [22, 31]. In addition, we assayed the loss of methylation when both RDR6- and Pol IV-RdDM are lost (in pol IV rdr6 double mutants) and found that this methylation level is slightly reduced compared to the pol IV single mutant (Fig. 1a), demonstrating that RDR6-RdDM plays a minor role when Pol IV-RdDM is mutated (see section below on RdDM compensation). In the ddm1 TE-active context, the overall CHH methylation level is reduced compared to the TE-silent context (Fig. 1a, replicate data in Additional file 4: Figure S3A) [19]. In addition, the ddm1 rdr6 double mutant shows lower CHH methylation compared to the ddm1 single mutant (Fig. 1a, replicate data in Additional file 4: Figure S3A), demonstrating a genome-wide role for RDR6-RdDM when TEs are reactivated.

Meta-plots of CHH methylation levels in TE-silent and TE-active contexts. a Average CHH methylation percentage across all DMRs identified in the TE-silent (top) or ddm1 TE-active (bottom) contexts. b Analysis of DMRs longer than 2 kb. c Alignment of all genes by their 5′ start and 3′ stop codons. d Alignment of all TEs by their 5′ and 3′ annotated boundaries. Orientation of the TE was determined using the TAIR10 TE annotation. e Alignment of all TEs longer than 2 kb. f Alignment of the transcriptionally competent subset of 2374 TEs. g Alignment of the transcriptionally competent TEs longer than 2 kb. Solid lines represent the 100 bp binned average CHH methylation percentages. The variation of individual element data points is represented as the transparent colored region around the solid lines (95 % confidence interval of the average)
In both the TE-silent and ddm1 TE-active contexts, loss of CHH methylation in pol IV mutants is reduced near the edge of the DMR and less so in the center of the DMR (Fig. 1a). To determine if this loss is due to Pol IV-RdDM functioning specifically at edges of long DMRs or if this effect is due to Pol IV-RdDM’s preference for short TE targets [19], we investigated only DMRs over 2 kb. We found that in the TE-silent context Pol IV-RdDM functions preferentially on long DMR edges, as the CHH methylation in pol IV mutants is lost more at the edge compared to the center of a >2 kb DMR (Fig. 1b). At the same time, we found the peak of high CHH methylation at the DMR edge (compared to the body of the DMR) in wt Col and ddm1 is a function of small DMRs in our analysis, as when only DMRs >2 kb are assayed, the CHH methylation values in wt Col or ddm1 are consistent over the length of the entire DMR (compare Fig. 1a to 1b, replicate data in Additional file 4: Figure S3A, B). Therefore, at least in the TE-silent context, Pol IV-RdDM targets short DMRs as well as the edges of long DMRs.
A DMR is a computationally identified feature that may span multiple TEs and genes or which may be as short as 4 bp. We found that analysis of the alignment of CHH methylation states of annotated genomic features (such as genes or TEs) was more informative than an analysis of the arbitrary edges of DMRs. For genes, we find that there is low average CHH methylation that is unaltered by Pol IV- or RDR6-RdDM, and we confirm that Pol IV-RdDM is responsible for gene-flanking methylation [22, 32], while RDR6-RdDM does not act near genes (Fig. 1c). For TEs, similar to our findings with DMRs, we find that rdr6 shows a CHH methylation loss only in the ddm1 TE-active context but not the TE-silent context (Fig. 1d, replicate data in Additional file 4: Figure S3D). We also observed that loss of CHH methylation in ddm1 rdr6 mutants occurs not specifically at the edge (as with Pol IV-RdDM at TE edges, see Fig. 1d), but rather acts over the length of the entire long TE and mostly from the TE internal region (Fig. 1e, replicate data in Additional file 4: Figure S3E). Interestingly, in the TE-active context Pol IV-RdDM acts like RDR6-RdDM throughout the length of the entire >2 kb TE (Fig. 1e). We observed this differential role of Pol IV-RdDM with DMRs as well (Fig. 1b) and these data demonstrate that the function of Pol IV-RdDM to reinforce silencing at short TEs and TE edges expands to silencing TE internal body coding regions when TEs are activated. In addition, for TEs >2 kb we find that the pol IV rdr6 double mutant has lower CHH methylation levels compared to either the rdr6 or pol IV mutants in either the TE-silent or TE-active context (Fig. 1d, e). This demonstrates that the finding on the single-locus level that some TEs are subject to both Pol IV- and RDR6-RdDM to direct full TE CHH methylation [12, 16] is also true on the genome-wide level.
To assess the role of Pol II expression on RdDM dynamics, we focused our analysis on transcriptionally competent TEs by identifying elements with direct evidence of mRNA production in ddm1 mutant plants (see “Methods”). For this set of 2374 TEs (7.6 % of all TEs) in the TE-silent context, we find that RDR6-RdDM does not function and Pol IV-RdDM’s role is reduced and primarily contributes to the edges of long TEs (Fig. 1f, g, replicate data in Additional file 4: Figure S3F, G). When this set of TEs is specifically transcribed, we find that RDR6-RdDM plays a larger role in TE methylation compared to Pol IV-RdDM, and this is pronounced in the internal regions of long TEs. Therefore, we conclude that RDR6-RdDM targets transcriptionally active TEs on the genome-wide level.
Dataset capture of both Dicer-dependent and Dicer-independent RdDM
Recent data have demonstrated that RdDM can occur through a Dicer-independent mechanism by which either transcribed or processed un-Diced RNAs of ~30–40 nt are trimmed into various small RNA sizes including 21–24 nt siRNAs [33–36]. This Dicer-independent production of small RNAs was shown to occur on both Pol IV and Pol II derived transcripts. While Dicer-dependent production generates specific siRNA size classes, Dicer-independent siRNA production creates small RNAs of all sizes, known as small RNA laddering [3: Figure S2C is shown as an arrow, while the TE shown in Fig. 3a is marked with an arrowhead. Genotypes are color-coded based on the methylation pathway (black = maintenance methylation (no RdDM), red = Pol IV-RdDM, blue = RDR6-RdDM, green = contributes to both Pol IV-RdDM and RDR6-RdDM)
Upon TE transcriptional activation, three RdDM mechanisms target genome-wide TE methylation
To characterize the methylation pathways that act on each TE genome-wide, we calculated the CHH methylation level for each of the annotated TE elements and fragments in the Arabidopsis genome (31,189). We were able to successfully cover and individually assay 29,252 (93.8 %) of all TEs, with the majority of TEs lost representing small high-copy TE fragments. We grouped TEs by their mechanism of CHH methylation: no CHH methylation, Pol IV-RdDM (dependent on Pol IV), RDR6-RdDM (dependent on RDR6), and maintenance methylation (not dependent on any RdDM) (see “Methods”) (Fig. 2e). The corresponding CG and CHG methylation analysis is shown in Additional file 6: Figure S5 and replicate data of CHH methylation patterns for key genotypes is shown in Additional file 4: Figure S3H. Similar to the TEs that have been individually investigated and determined to be targets of RDR6-RdDM [12, 16], we found that both RDR6-RdDM and Pol IV-RdDM can target the same TE, providing a distinct co-regulated category (Fig. 2e). In addition, we identified a category of TEs that are methylated by a new pathway of DCL3-dependent 24 nt siRNAs which are not produced from Pol IV, a pathway we refer to as DCL3-RdDM (see below).
Genome-wide distribution of TE CHH methylation in the TE-silent context demonstrates that roughly one-third of TEs do not have CHH methylation, roughly one-third of TEs are not going through RdDM and are subject to only maintenance CHH methylation via CMT2, and roughly one-third are regulated by Pol IV-RdDM (Fig. 2e). This confirms that when TEs are silenced, maintenance methylation and Pol IV-RdDM are the major pathways that mutually exclusively target TE CHH methylation [7]. We find that pol IV and rdr2 mutants have less TE CHH methylation than dcl3 mutants (Fig. 2e), supporting the Dicer-independent function of Pol IV/RDR2-derived siRNAs in RdDM [2




