
Abstract
Colorectal cancer liver metastasis (CRLM) is one of the leading causes of death among patients with colorectal cancer (CRC). Although immunotherapy has demonstrated encouraging outcomes in CRC, its benefits are minimal in CRLM. The complex immune landscape of the hepatic tumour microenvironment is essential for the development of a premetastatic niche and for the colonisation and metastasis of CRC cells; thus, an in-depth understanding of these mechanisms can provide effective immunotherapeutic targets for CRLM. This review summarises recent studies on the immune landscape of the tumour microenvironment of CRLM and highlights therapeutic prospects for targeting the suppressive immune microenvironment of CRLM.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is the third most common cancer and the second leading cause of cancer-related mortality worldwide [1]. Although colonoscopy screening has become popularised, the morbidity and mortality of CRC remain high among men [2]. Early-stage CRC is eligible for curative treatment [3]; however, 25–50% of patients with early-stage disease progress to metastatic disease [4]. The liver is the most frequent site of metastasis in patients with CRC [5]. Blood draining from the gastrointestinal tract enters the liver through the portal vein, which promotes the dissemination of CRC into the liver [6, 7]. Approximately 15–25% of patients with CRC have synchronous liver metastasis (LM) [8, 9], and 18–25% of patients with CRC may eventually develop metachronous LM within 5 years of the initial diagnosis [10]. The 5-year survival rate dramatically declines when the local disease develops into metastasis [11, 12]. Therefore, LM has been used as a prognostic marker for CRC. Despite the development of surgical techniques and targeted therapy, the prognosis of colorectal liver metastasis (CRLM) remains poor [13].
The tumour microenvironment (TME) is composed of cancerous and noncancerous cells, including fibroblasts, endothelial cells and immune cells, as well as noncellular components such as the extracellular matrix (ECM), cytokines, growth factors and extracellular vesicles (EVs) [14, 47, 52, 53]. (1) Microvascular phase: Liver-infiltrating CRC cells that are trapped in sinusoidal vessels are killed via phagocytosis by Kupffer cells (KCs) and natural killer (NK) cell-mediated antitumour cytotoxicity [54, 55]; they may also remain alive by esca** cytotoxic effects and adhering to LSECs [53], which facilitates CRC migration into the space of Disse to avoid immune killing. (2) Extravasation and preangiogenic phase: CRC cells relocate to the space of Disse, thus recruiting stromal cells, including hepatic stellate cells (HSCs) that are responsible for the secretion of fibronectin and collagen to form a framework for neovascularization [56, 57] and portal tract fibroblasts, which generate IL-8 to promote invasion and angiogenesis [58]. (3) Angiogenic phase: After LSECs are activated and co-opted to the tumour–liver interface, activated HSC-derived vascular endothelial growth factor (VEGF) induces the formation of intrametastatic vessels, which appear to be continuous with sinusoidal vessels [59]. Various immunosuppressive cells, such as immunosuppressive regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs) and macrophages, are activated to form an immunosuppressive microenvironment, which promotes the development of CRLM. (4) Growth phase: CRC cells acquire adequate blood supply and proliferate rapidly under the ‘protection’ of intrinsic hepatic immune tolerance and the immunosuppressive microenvironment, eventually forming a detectable metastatic tumour in clinical settings [47]. Therefore, the targeting of angiogenesis and the transformation of the immunosuppressive microenvironment into an immune-effective microenvironment are prospective therapeutic strategies for CRLM. Moreover, an understanding of the immune microenvironment of the liver may help to develop effective immunotherapeutic approaches.
Immune landscape of the TME in CRLM
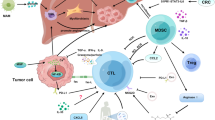
The homeostasis maintained by organ innate resistance in the liver is attributed to various highly specialised resident cells and all types of immune cells [21, 60,61,62]. Each of these cells not only helps to balance protein, lipid and glucose metabolism but also orchestrates immune responses and oncogenesis [63,64,65,66,67,68]. The considerable inflow of antigens shapes the unique immune microenvironment of the liver to harmonise immune activation and immune tolerance [69, 70]. In the early stage of CRLM, the abovementioned cells act as defenders to destroy disseminated cancer cells. Specifically, LSECs arrest cancer cells, whereas KCs phagocytose and release tumour-killing cytokines. Additionally, APCs present antigens to T cells and transform them to effector T cells, which is strengthened by CD4+ T cells. Cytokines released from natural killer T (NKT) cells and M1 macrophages protect against cancer cells. However, when cancer cells escape the immune system, effector T cells are rendered dysfunctional by immune checkpoints, whereas cancer cells migrate into the space of Disse by adhering to LSECs. Treg cells impair the antigen-presenting activity of dendritic cells (DCs). Moreover, HSCs are activated to promote ECM remodelling, and M2 macrophages produce MMPs to regulate this process. Tumour-associated neutrophils (TANs) extrude chromatin fibres and form neutrophil extracellular traps (NETs), which trap CRC cells in the liver and eventually promote their invasive and metastatic capabilities. Furthermore, CRC cells adhere to hepatocytes and induce the release of serum amyloid A1 and A2 (SAA) and insulin-like growth factor-I (IGF-I) from hepatocytes, thereby making the liver a primary target for CRC metastasis (Fig. 2).

A schematic representation of the immune landscape of the TME in CRLM. GZM, granzyme; PRF, perforin; CTL, cytotoxic T lymphocyte; BA, bile acid; ex CD8+ T cell, exhausted CD8+ T cell; NET, neutrophil extracellular trap; CCRK, cell cycle-related kinase; sLewis-x, sialyl Lewis-x; CTLA-4, cytotoxic T lymphocyte antigen-4; SAA, serum amyloid A1 and A2; IGF-I, insulin-like growth factor-I. ① LSECs and NK cells produce IFNγ to upregulate functional Fas and induce apoptosis of cancer cells; PRF and GZM released from NK cells kill cancer cells. ② Disseminated CRC cells are phagocytosed by KCs along with the release of TNF-α, IL-1α and IL-1β. ③ APCs present neoantigens to CD8+ T cells, thus inducing the rapid proliferation of CD8+ T cells and their differentiation into CTLs. ④ CTLs secrete PRF and GZM, as assisted by IFNγ and TNF-α produced by Th1 cells to kill cancer cells. ⑤ LSECs are regulated by gut microbiota-modified bile acids to secret CXCL16, thus recruiting NKT cells to fight cancer cells. ⑥ M1 macrophages directly kill cancer cells by releasing cytotoxic ROS, NO and IL-12. ⑦ The function of cytotoxic CD8+ T cells is impeded due to the interplay between PD-L1 and PD-1. ⑧ The interaction between E-selectin and sialyl Lewis-x promotes the adhesion of CRC cells to LSECs. ⑨ Treg cells bind to APCs via the interaction between CTLA-4 and CD80/86 and produce TGF-β and IL-10 to suppress the activation of CTLs. ⑩ MDSCs, which are recruited by CXCL1 secreted from CRC cells, induce the activation of Treg cells to impair the antigen-presenting activity of DC cells. ⑪ M2 macrophages produce IL-10, TGF-β and MMP to regulate matrix remodelling. ⑫ As induced by TGF-β secreted from KCs, HSCs are transformed to aHSCs and release TGF-β to promote ECM remodelling. ⑬ Lactic acid causes NK cell apoptosis by downregulating their intracellular pH. ⑭ TANs release CCL2 and CCL17 to recruit CCR2+ M2 macrophages and CCR4+ Treg cells. ⑮ As induced by IL-8, NETs trap CRC cells in the liver. ⑯ Hepatocyte-derived CCRK increases CXCL1 production to recruit PMN-MDSCs, thereby impairing NKT cell-mediated immunosurveillance. ⑰ As mediated by integrins and desmosomes, CRC cells adhere to hepatocytes, thus inducing the release of SAA and IGF-I from hepatocytes
Interaction of resident liver cells with cancer cells
Liver sinusoidal endothelial cells
LSECs perform important physiological and immunological functions, including filtration, endocytosis and antigen presentation [71,72,73]. As a selective barrier, LSECs allow for the entry of molecules such as plasma proteins, drugs, small chylomicron remnants, exosomes and smaller viruses (< 200 nm) into the space of Disse; however, they do not allow the entry of cells [74,75,76]. The mannose receptor, scavenger receptor and Fc-γ receptor IIb2 efficiently facilitate the clearance and degradation of blood-borne macromolecules by LSECs to perform endocytosis and scavenging functions [77, 78]. Additionally, LSECs exert antigen-presenting functions mediated by the mannose receptor and scavenger receptor, which mainly reshape the immunosuppressive microenvironment in the liver. However, LSECs can dampen effector immune responses. Specifically,, antigen presentation by LSECs mainly induces the differentiation of CD4+ T cells into Treg cells to promote the development of an immune-tolerant TME in the liver [44, 79]. In contrast, LSECs mainly drive a tolerogenic response mediated by an increase in the levels of coinhibitory PD-L1 that interacts with PD-1 to induce CD8+ T-cell dysfunction [80].
LSECs play a dual role in advancing tumorigenesis. When disseminated CRC cells enter the sinusoids, they are entrapped by LSECs and are either destroyed due to mechanical stress, phagocytosed by KCs or killed by perforin (PRF)/granzyme (GZM) from NK cells. LSECs and NK cells release interferon-gamma (IFNγ) and nitric oxide (NO) to upregulate Fas and induce apoptosis of cancer cells via the Fas–FasL pathway [81]. Recent studies have highlighted the fact that LSECs are influenced by gut microbiota-modified bile acids to secrete CXCL16, which recruits NKT cells to fight primary and metastatic liver tumours [82]. However, the anticancer proinflammatory response results in the high expression of vascular adhesion factors such as E-selectin, VCAM-1 and ICAM-1 on LSECs, thus leading to the susceptibility of LSECs to adhesion by cancer cells with the help of sialyl Lewis-x, PSGL-1 and ESL-1 [83,84,85]. Cancer cells can escape from the destruction of the initial assault through counterreceptor communication, after which they migrate into the space of Disse, where they are protected from the cytotoxic effects of KCs and NK cells [86]. A novel adhesion molecule known as LSECtin mediates the communication between activated T cells and LSECs [87, 88] to inhibit the tumour-killing effects of T cells; in addition, it facilitates adhesion and migration of CRC cells to the liver [89]. In multiple experimental LM models, melittin nanoparticles have been demonstrated to induce the activation of LSECs to reverse the hepatic immunological environment to the activated state, which recruits NK and CD8+ T cells and suppresses LM [90].
Due to the fact that LSECs induce a suppressive immune microenvironment in the liver and assist in the growth of disseminated cancer cells, the targeting of LSECs to modulate the hepatic immune microenvironment may be a novel approach to the management of LM in the future. For example, the abundance of beneficial gut organisms that optimise the metabolism and immunity of the liver can be enhanced by modulating the action of LSECs for the effective treatment of CRLM.
Kupffer cells
KCs, which are the resident macrophages in the liver, serve as a crucial part of the innate immune response, which is the first line of defence of the liver [91]. Localised in the hepatic sinusoid, KCs can recognise all types of antigens (such as immune complexes, senescent cells and cancer cells) from the portal or arterial circulation and exert anti-inflammatory effects to prevent the entry of gut-derived substances into the hepatic sinusoid [92, 93]. In the early stage of CRLM, the adherence of disseminated cancer cells to KCs prompts KCs to capture and phagocytose the cancer cells and release TNF-α, interleukin-1α (IL-1α) and IL-1β, thus reducing the metastasis of colon cancer cells to the liver [94, 95]. The innate receptor Dectin-2 on KCs promotes the phagocytosis and elimination of disseminated CRC cells to resist metastasis [96].
Although KCs mainly play a tumoricidal role in the early stages of metastasis, they also play a vital role in hepatic carcinogenesis [97]. KCs activate and expand FOXP3+CD4+ Treg cells through antigen presentation and induce tolerance by upregulating the inhibitory marker PD-L1, thereby resulting in the formation of an immune-tolerant environment to achieve homeostasis [23]. Moreover, HSCs are activated and produce fibronectin induced by KC-derived TGF-β, thus recruiting bone marrow-derived macrophages and neutrophils to form a favourable environment [53]. The corelease of TGF-β, fibronectin, EGF, VEGF and matrix metalloproteinases (MMP-2, MMP-9 and MMP-13) from KCs and HSCs leads to ECM remodelling, angiogenesis and cancer progression [98], which is augmented by the absorption of pancreatic ductal adenocarcinoma (PDAC)-derived exosomes by KCs in a PDAC model [99]. However, CRC-derived exosomal angiopoietin-like protein 1 (ANGPTL1) shuttles to KCs to decrease the expression of MMP-9, which subsequently reduces LM and inhibits vascular leakage mediated via the suppression of the JAK2-STAT3 signalling pathway [100]. In addition, KCs can phagocytose EV-packaged miR-135a-5p, thus mediating immunosuppression and facilitating the development of a premetastatic niche (PMN) in patients with CRLM [101].
It has been reported that a novel immunotherapy strategy by using bacterial genetic modification induces the reprogramming of KCs, which augments the phagocytic ability of cancer cells and strengthens the cytotoxic killing capacity of T cells to suppress LM [102]. An understanding of the functional role of KCs in CRLM may help to identify potential therapeutic targets and to develop novel therapeutic strategies, such as nanoparticle-mediated noncoding RNA-based therapy and bacterial treatment to reprogram the function of KCs. However, further research is required to identify the underlying mechanism and potential for application.
Hepatic stellate cells
As a resident nonparenchymal liver cell population, HSCs contribute to liver fibrosis and cancer development [67, 103]. HSCs maintain homeostasis in the liver by regulating the ECM, immune tolerance and inflammatory responses; additionally, they play a significant role in the colonisation and metastasis of cancer cells [104,105,106].
TGF-β is an important regulator of HSCs in the hepatic microenvironment. Its high expression blocks the initiation of CD4+ Th1 cells and weakens cytotoxic responses, thus facilitating LM and leading to a poor prognosis [107, 108]. TGF-β can induce the transformation of HSCs into a fibroblast-like (spindle-like and spread) phenotype (known as activated HSCs [aHSCs]) to promote ECM remodelling [98]. In addition, aHSCs play a vital role in secondary or primary hepatocellular carcinoma [109,110,111]. aHSCs can lead to hepatic fibrosis and portal hypertension, thus contributing to hepatocarcinogenesis and metastasis [109]. In a previous study, we demonstrated that CRC-derived exosomal miR-181a-5p facilitates CRLM by activating HSCs [112]. In addition, aHSCs engulf disease-associated lymphocytes, including CD8+ T, CD4+ T and NK cells, through cell adhesion [113]. As key cells involved in pro-tumour angiogenesis, HSCs have been demonstrated to upregulate fibroblast activation protein alpha (FAPα) and increase CXCL5 secretion, as regulated by cancer cell-secreted fibroblast growth factor-binding protein 1 (FGFBP1). This mechanism stimulates epithelial–mesenchymal transition (EMT) and induces vessel co-option that results in bevacizumab resistance in CRLM models [114]. Therefore, HSCs play an important role in sha** the immune microenvironment of the liver and in inducing resistance to antiangiogenic therapy. The targeting of HSCs expressing specific molecules (such as FAPα) to modulate the immune microenvironment of CRLM represents a beneficial strategy for strengthening the antitumour effects of immune cells and for effectively overcoming drug resistance.
Hepatocytes
Hepatocytes play an essential role in inducing an immune-tolerant TME, which is required for the implantation of disseminated cancer cells. Hepatocyte-mediated cross-presentation of soluble antigens can induce tolerance of antigen-specific CD8+ T cells [115]. After extravasation, disseminated CRC cells can deeply penetrate into the hepatocyte plate, where they proliferate and form metastatic foci. The adhesion of CRC cells to hepatocytes is considered an essential step in the formation of LM [116], which is mediated by integrins [116] or desmosomes [117]. The strongly expressed integrin subunit αvβ5 mediates cell migration and LM in CRC, and its effects are enhanced by hepatocyte-derived heregulin [118]. Hepatocyte-derived SAA can facilitate the development of LM and is highly expressed in patients with CRC. Mechanistically, hepatocytes promote LM by activating IL-6–STAT3 signalling and inducing SAA overexpression, thereby resha** the hepatic TME to facilitate the formation of a PMN in the liver [119]. Moreover, IGF-I can affect cancer growth and metastasis. The inhibition of IGF-1 released from hepatocytes reduces CRLM in mice [120]. A novel IGF-targeting protein (IGF-Trap) has been demonstrated to markedly block CRLM in experimental models to compensate for the function of the impaired insulin receptor system, thus inducing tumour cell apoptosis and reducing angiogenesis [121].
Altogether, the interplay between resident liver cells and cancer cells contributes to the progression and spread of CRC (Table 1). A better understanding of the communication between CRC cells and the hepatic TME may facilitate the development of new combination therapies for the efficient management of CRLM.
Immune cells contributing to the hepatic immune microenvironment of CRLM
CD4+ T cells
CD4+ T cells are essential in the defence against tumours because they regulate the activity of CD8+ T cells and influence the outcome of antitumour responses [122]. The classical effector CD4+ T helper 1 (Th1) and T helper 2 (Th2) subsets elicit important antitumour immune responses. Specifically, Th1 cells produce cytokines such as IFNγ and TNF-α, thus leading to cell-mediated killing, whereas Th2 cells secrete IL-4, which assists in the activation of humoral immunity [123]. In addition, CD4+ T cells can differentiate into new subsets, such as Th9 cells, Th17 cells and FOXP3+ Treg cells. Moreover, the role of Th17 cells in cancer is controversial [124, 125]. The low proportion of Th1 cells and high proportion of Th17 cells in liver metastatic tissue indicate a poor prognosis in patients with CRLM [126], which is consistent with the condition of patients with CRC [127]. Given the immunosuppressive activity of CD4+ T cells, we mainly focused on FOXP3+CD4+ Treg cells in this review.
One of the characteristics of LM progression is the high infiltration of FOXP3+ Treg cells [152]. Moreover, exosomal circCCAR1 expressed by cancer cells communicates with CD8+ T cells to impede the degradation of PD1, thus promoting the exhaustion of CD8+ T cells in the liver [153]. In addition, the high expression of MGP in cancer cells from both the primary CRC or LM sites increases intracellular Ca2+ to boost NF-κB phosphorylation, which mediates PD-L1 upregulation in CRC cells, thus promoting CD8+ T-cell dysfunction [154]. Furthermore, the induction of T cells to differentiate into regulatory cells is mediated by IL-10 release from LSECs, which are prone to activating the regulatory pathway of CD4+ T cells to FOXP3+CD4+ Treg cells [145].
Recent studies on the apoptosis of CD8+ T cells have demonstrated that tumour-associated macrophages (TAMs) induce apoptosis of CD8+ T cells and impair cytotoxic functions by reducing the expression of granzyme B and perforin in the liver [155]. The mechanism for this effect involves the fact that activated CD8+ T cells experience apoptotic cell death by the Fas–FasL pathway, as mediated by TAMs within the liver, which induces a decrease in activated T cells and transforms the hepatic immune microenvironment in CRLM [25] (Fig. 3C).
Given that cancer-reactive CTLs play a central role in cancer immunity, it is important to reactivate CD8+ T cells to suppress the progression and metastasis of CRC. It has been reported that hyper-IL-15, IL-15 and the sushi domain of the IL-15 receptor α chain augment the cytotoxic functions of CD8+ T and NK cells, which may be a prospective therapy to reactivate CD8+ T cells and recover their anticancer ability to manage CRLM [156].
Tumour-associated macrophages
As multifunctional APCs, macrophages are critical mediators of tumour immunity [157]. Macrophages present exogenous antigens to T cells through MHC-I and MHC-II aided by costimulatory signals, inhibitory signals or other cytokine signals to regulate T-cell activation [158]. Macrophages that infiltrate malignant tissues are known as TAMs.
With inherent plasticity and polarising characteristics, TAMs are conventionally categorised into two subtypes: M1 and M2 macrophages [159, 160]. M1 macrophages suppress cancer growth by releasing cytotoxic reactive oxygen species (ROS), NO and IL-12, which can directly kill cancer cells [161]. However, M2 macrophages induce the formation of an immunosuppressive TME by secreting cytokines, including IL-10, TGF-β, CCL17 and CCL22 [157, 162]. Due to their poor ability to present cancer antigens, M2 macrophages undermine Th1 adaptive immunity [163]. In addition, M2 macrophages produce MMPs to regulate matrix remodelling, thus facilitating the invasion and metastasis of cancer [164]. In CRC, the expanding liver metastatic tumour is rich in TAMs (primarily M2 macrophages) [157, 171]. Mechanistically, CRC-derived CTHRC1 interacts with the TGF-β receptor in macrophages to activate TGF-β signalling to promote CRLM.
With the development of single-cell profiling, TAMs have been classified as C1QC+ TAMs, SPP1+ TAMs and MRC1+CCL18+ TAMs [151, 165, 172]. In a previous study, single-cell analysis showed that MRC1+CCL18+ macrophages and SPP1+ macrophages are the predominant M2 cell subsets in liver metastatic tissue [151]. Consistently, the presence of SPP1+ macrophages in liver metastatic tissue was reported in a study by Liu et al. Therefore, SPP1+ macrophages may be a potential culprit in CRLM. Moreover, MRC1+CCL18+ macrophages infiltrating liver metastatic tissue exhibit high metabolic activities, thus suggesting that they may promote LM through metabolic pathways [151]. SPP1+ macrophages are found in mesenteric lymph nodes with metastasis but not in mesenteric lymph nodes without metastasis, thus indicating that SPP1+ macrophages play a role in facilitating the expansion of disseminated cancer cells [165]. Furthermore, it has been reported that in microsatellite-stable (MSS) CRC, SPP1+ macrophages and fibroblasts communicate very closely via the ligand‒receptor pathway, which may help to shape an immunosuppressive TME in the liver [173]. However, more studies are required to understand the mechanisms by which different subtypes of macrophages promote LM.
Overall, as the leading tumour-infiltrating immune cells in the TME [174,175,176], TAMs play a critical role in the progression and metastasis of CRC. Their high proportion is closely related to a worse prognosis [165, 177]. Specific subsets of macrophages, including MRC1+CCL18+ and SPP1+ macrophages, may serve as potential therapeutic targets for CRLM.
Myeloid-derived suppressor cells
MDSCs are one of the key contributors to the formation of an immunosuppressive TME in the liver [178]. They mediate immune evasion by inducing the production of Treg cells [179], thus inhibiting NK cell function [180] and impairing the antigen-presenting activity of DCs [181]. In addition, MDSCs facilitate cancer progression and metastasis in a nonimmune manner by producing MMP-9, which is a primary regulator of EMT [182], as well as VEGF, in order to promote TME remodelling and angiogenesis [20]. MDSCs are mainly classified as granulocytic or polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). The phenotypic and molecular features of these subtypes are difficult to identify [183]. Furthermore, the accumulation of MDSCs is one of the most dominant immunological features of CRC and is associated with disease progression and metastasis [184, 185].
MDSCs facilitate the formation of a PMN and the metastatic colonisation of CRC [186, 187]. Clinically, the high expression of CCL15 in patients with CRC results in the recruitment of more CCR1+ MDSCs, which is associated with the loss of SMAD4 (which is a TGFβ-relevant transcription factor) and promotes CRLM [188, 189]. In an orthotopic mouse model of CRC, CXCR2-expressing MDSCs are recruited from the circulatory system to the liver by CXCL1 secreted from CRC cells in the premetastatic liver, which facilitates the growth of disseminated CRC and its metastasis to the liver [187]. Mechanistically, sphingosine-1-phosphate receptor 1 (S1PR1)–STAT3 signalling in CRC cells results in the production of IL-6 to induce the activation of S1PR1 and p-STAT3 in MDSCs, thus leading to the formation of a PMN in the liver to promote CRLM [190]. Zeng et al. reported that the overexpression of hepatocyte-derived cell cycle-related kinase (CCRK) increases CXCL1 production to recruit PMN-MDSCs, thereby impairing NKT cell-mediated immunosurveillance, which dramatically promotes the metastasis of CRC cells to the liver [191]. M-MDSC-produced CCL7 binds to CCR2 on micrometastatic cells and stimulates the JAK/STAT3 pathway to activate dormant cells, thereby promoting the progression of CRLM [192]. Moreover, the inhibition of CCL7 may represent a potential strategy for preventing recurrent CRLM.
However, it is difficult to target MDSCs because they do not have a specific phenotype that differs from other mature granulocytes. Therefore, further research is required to identify therapeutic targets.
Natural killer cells
Under physiological conditions, NK cells are enriched in the liver and contribute to defending against infection and eliminating cancer cells [69]. After NK cells encounter cancer cells and are activated, they release PRF and GZM, thus leading to osmotic lysis and apoptosis of cancer cells [193]. Additionally, NK cells can directly kill target cells via the expression of TNF-related apoptosis-inducing ligand and FasL [194]. NK cells function in tumour immunosurveillance and elicit inflammatory responses by producing cytokines and chemokines [195].
NK cells can eliminate disseminated cancer cells to control metastasis [196]. A high proportion of NK cells indicates a good prognosis in patients with CRLM [197]. However, in the highly glycolytic environment of CRLMs, lactic acid causes the apoptosis of NK cells by downregulating their intracellular pH [198]. In addition, MDSCs attenuate the immunoreaction of NK cells by releasing NO, which interferes with FcR-mediated functions of NK cells, such as antibody-dependent cellular cytotoxicity (ADCC) and cytokine generation [199]. In a previous study on murine models of CRLM, compared with conventional NK (cNK) cells, liver-resident natural killer (LrNK) cells had a high expression of RORα, which is required to maintain LrNK cells but has no impact on cNK cells. The conditional knockout of Rorα aggravated CRLM, thus indicating that RORα is required for LrNK cell-mediated antitumour immunity. However, the RORα agonist SR1078 restrained CRLM [200]. Clinically, LrNK cells are significantly depleted in CRLM due to the accumulation of tumour-derived lactate, thus resulting in mitochondrial dysfunction and apoptosis of NK cells [198]. The targeting of lactate in the TME may restore the tumour-killing effects of NK cells and benefit patients with CRLM. Altogether, LrNK cells exert a great antitumour impact on CRLM and are closely related to prognosis; therefore, they may be qualified as specific therapeutic targets for CRLM.
Dendritic cells
Dendritic cells are the classical APCs that exert considerable influence in triggering antigen-specific immune responses and inducing immune tolerance [201,202,203,204]. The antigen-presenting function of conventional DCs (cDCs) is important for the antitumour response of effector T cells [205,206,207]. Efficient antigen presentation increases the polarisation of CD4+ Th1 cells and the activation of CD8+ T cells [208, 209].
DCs are heterogeneous and exhibit different characteristics. Compared with plasmacytoid DCs (pDCs), cDCs can more efficiently initiate an immune response against cancer cells [210]. In ICB-treated mouse models of orthotopic pMMR CRLM, the proportion of activated CD8+ T cells, CD4+ T cells and cDCs is lower in metastatic tumours than in subcutaneous tumours [211]. Liver-derived pDCs have a poor capability to stimulate the proliferation of T cells, thus resulting in anergy of effector T cells and immune suppression to maintain inherent liver tolerogenicity [212, 213]. Moreover, liver-resident regulatory DCs differentiated from bone marrow-derived progenitors secrete high levels of IL-10 but low levels of IL-12, thereby inhibiting effective T-cell function to maintain liver tolerance [214]. A subset of cDCs in CRLM identified as DC3s induces a proinflammatory phenotype and is correlated with a poor prognosis [165]. DC3s may be considered as a promising target for improving the therapeutic outcome of immunotherapy in CRLM. Further investigations are required to elucidate the mechanism by which DC3s promote CRLM.
Tumour-associated neutrophils
Similar to TAMs, TANs play a dual role in cancer progression by both promoting and inhibiting the growth and metastasis of cancer [215]. Specifically, TANs facilitate activated T-cell immune reactions by presenting antigens and releasing IL-18 to induce the activation of NK cells [216]. In contrast, TANs release CCL2 and CCL17 to recruit CCR2+ M2 macrophages and CCR4+ Treg cells, which shape a suppressive TME in the liver, thus promoting the progression and metastasis of cancer [217]. Additionally, TANs produce MMP-9 and neutrophil elastase to promote the extravasation of cancer cells and drive disseminated cancer cells to metastasise [218].
The accumulation of TANs has been demonstrated to be necessary for the formation of an omental PMN in orthotopic ovarian cancer models [219]. Ovarian cancer induces neutrophils to form NETs, which trap ovarian cancer cells and facilitate their implantation on the omentum [219]. Therefore, NETs can promote the metastasis of ovarian cancer. Several in vivo and in vitro studies on CRC have reported that the formation of NETs is enhanced by cancer-derived IL-8. These NETs can trap CRC cells in the liver and promote their invasive and metastatic capabilities [220,221,222]. In addition, anterior gradient-2 (AGR2) released from TANs can promote metastasis in murine models of CRLM. TAN–CRC cell crosstalk between TAN-derived AGR2 and CRC-derived TGF-β1 is considered the primary driver of CRLM [223]. Collectively, TAN is an effective potential target for the treatment of CRLM. However, further investigations are required to explore and develop TAN-based therapeutic strategies for CRLM.
In conclusion, immune cells involved in CRLM shape the susceptible suppressive immune microenvironment for tumour invasion and metastasis in CRC (Table 2). Immunotherapeutic strategies that can reverse the immunosuppressive microenvironment or strengthen effector immunity may be effective against CRLM.
Extracellular vesicles in the immune microenvironment of CRLM
EVs refer to various nanosized vesicles with membrane structures released by cells [224, 225]. According to their diameter and the mechanisms of biogenesis, they are classified into three subgroups (exosomes, microvesicles and apoptotic bodies). Exosomes have attracted substantial interest and have been widely investigated in recent years [226]. EVs carry bioactive molecules such as nucleic acids, proteins and lipids for intercellular delivery and facilitate intercellular communication [227, 228]. They are an important aspect of the immune microenvironment. CRC-derived EVs in the immune microenvironment facilitate the relocation and aggression of CRC, which contributes to LM [229,230,231,232]. In previous studies, we elucidated the molecular mechanisms underlying the involvement of EVs in the formation of a hepatic PMN and the metastasis of CRC, and we also identified promising functional biomarkers for CRC [35, 112, 233, 234], thus indicating that EVs may serve as a therapeutic target and a prognostic and diagnostic biomarker for CRLM (Fig. 4).

Schematic diagram depicting extracellular vesicles in the immune microenvironment of CRLM. Various cells in the hepatic immune microenvironment interact with CRC cells via extracellular vesicles to form a sophisticated immunosuppressive microenvironment that contributes to CRLM. The different pathways are indicated by different coloured arrows. ① CRC-derived hypoxia-induced exosomal miR-135a-5p is phagocytosed by KCs, thus blocking CD30-mediated CD4+ T-cell activation and promoting cell adhesion. ② Highly mCRC cells produce EV-packaged miR-181a-5p that activates HSCs. aHSCs release CCL20, which interacts with CCR6 expressed on CRC cells and activates CRC cells to promote the release of EV-packaged miR-181a-5p, thus contributing to resha** the hepatic TME and forming a PMN; CRC-derived exosomal HSPC111 promotes the activation of HSCs, thus leading to the upregulation of CXCL5, which targets CRC-expressed CXCR2, increases the secretion of exosomal HSPC111 from CRC cells and promotes CRLM. ③ M2 macrophages release exosomal miR-21-5p and miR-155-5p, after which they shuttle into CRC cells, which contributes to the migration and invasion of CRC. ④ Exosomal miR-934 secreted from CRC cells induces M2 macrophage polarisation to promote CRLM. M2 macrophages release CXCL13, which interacts with CXCR5 in CRC cells and promotes the transcription of miR-934. ⑤ CRC cells secrete exosomal miR-25-3p to stimulate endothelial cells, thus leading to vascular leakage and vasculogenesis
Several studies have validated the pivotal role of EVs in the formation of a hepatic PMN. After being phagocytosed by KCs, CRC-derived EV-packaged miR-135a-5p can inhibit CD30-induced activation of CD4+ T cells and promote cell adhesion, which facilitates the development of a PMN for CRLM [101]. In a previous study, we reported that EV-packaged miR-181a-5p secreted by CRC cells activates HSCs. The interaction between CCL20 released from aHSCs and CCR6 expressed on CRC cells activates CRC cells to promote the release of exosomal miR-181a-5p, which generates a positive feedback loop to reshape the hepatic TME and form a PMN [112]. In mouse models of CRLM, endothelial cells stimulated by CRC-derived exosomal miR-25-3p can lead to vascular leakage and vasculogenesis, thus contributing to the formation of a PMN and enhancing CRLM [235].
EVs play an important role in intercellular communication, such as between CRC cells and other cells, in CRLM. M2 macrophage-derived exosomes can deliver miR-21-5p and miR-155-5p to CRC cells. Additionally, miR-21-5p and miR-155-5p are internalised by CRC cells and targeted to the BRG1 coding sequence, thus leading to a decrease in the expression of BRG1 in CRC cells and contributing to the migration and invasion of CRC [236]. In a previous study, we demonstrated that exosomal miR-934 in the immune microenvironment induces M2 macrophage polarisation to promote CRLM. Additionally, CXCL13 released by M2 macrophages interacts with CXCR5 on CRC cells to promote the transcription of miR-934 [35]. CRC-derived exosomal HSPC111 can promote the activation of HSCs, thus leading to the upregulation of CXCL5, which targets CXCR2 expressed on CRC cells, increases the release of exosomal HSPC111 from CRC cells and promotes CRLM [237].
Altogether, as coordinators of intercellular communication in the dynamic network of the TME, EVs are responsible for the progression and metastasis of cancer [238] and can serve as noninvasive markers for the screening and management of CRLM [239, 240]. A better understanding of the regulatory mechanisms of EVs can help to generate antitumour responses and design efficient EV-based diagnostic and therapeutic strategies for CRLM.
Therapies for CRLM
Surgical resection
At present, surgical resection is an effective therapeutic option for resectable CRLM [241]. The two commonly used surgical strategies include simultaneous resection and delayed resection. The results of existing studies on the selection of simultaneous or delayed resection are inconsistent [242,243,244]. The 5-year survival rate of patients with CRLM after resection can be improved to 50% [241]. However, only 10–30% of patients with localised LM are eligible for resection after diagnosis [245]. Moreover, 52% of patients develop postoperative recurrence of CRLM [246], thus resulting in a high mortality rate. Therefore, there is a need to explore novel therapeutic modalities for CRLM.
Systemic and conversion therapies
Systemic therapy is a more favourable treatment option for nonresectable CRLM. In addition to improving the quality of life and prolonging survival, effective systemic therapy can transform unresectable lesions into resectable lesions, which is known as conversion therapy [247]. According to the guidelines recommended by the National Comprehensive Cancer Network (NCCN) [248], first-line chemotherapy regimens for patients eligible for intensive therapy are FOLFOX (5-fluorouracil combined with leucovorin plus oxaliplatin), CAPEOX (combination of capecitabine and oxaliplatin), FOLFIRI (5-fluorouracil plus leucovorin and irinotecan) and injectable 5-fluorouracil/leucovorin or capecitabine. The use of FOLFOXIRI (5-fluorouracil combined with leucovorin, oxaliplatin and irinotecan) in conversion therapy may maximise tumour shrinkage and improve the eventual outcomes of surgery in patients with potentially resectable CRLM. Effective conversion therapy can allow for 12.5% of patients with unresectable CRLM to undergo liver resection, thus resulting in improved survival rates [249]. However, several adverse effects are associated with this regimen, which should be carefully considered.
Chemotherapy combined with targeted therapy can yield a better outcome for patients who are tolerant to aggressive therapy [250]. Drugs targeting epithelial growth factor receptor (EGFR) and VEGF are commonly used in combination with chemotherapeutic drugs. A phase III trial demonstrated that cetuximab and panitumumab (which are monoclonal antibodies against EGFR) can suppress the downstream signalling pathways of EGFR to effectively inhibit disease progression and provide clinical benefits to patients with mCRC [251]. However, cetuximab and panitumumab are only indicated in patients with wild-type RAS/BRAF [248]. Bevacizumab targets VEGF and plays a significant role in antiangiogenesis [5).
Various vaccine strategies have been designed, including whole tumour cell-based, protein- or peptide-based and DC-based vaccines [297,298,299]. In a randomised clinical trial, patients with colon cancer who received an autologous cancer cell-based vaccine had a significantly longer recurrence-free interval (p = 0.011) and recurrence-free survival (p = 0.032); however, disease-specific survival and OS showed no improvement [300]. Based on this foundation, further clinical studies (Clinicaltrials.gov Identifier: NCT00016133, NCT02448173) have been conducted to examine the protective effects of the autologous cancer vaccine against tumour recurrence after colon cancer surgery [301]. DC-based vaccines are powerful contributors to antigen presentation and the initiation of antitumour immunity [302,303,304]; however, the development of DC-based vaccines for CRC is currently at an early stage. In a preclinical murine model, effective immunotherapy using tumour-associated antigen-loaded cDC-based vaccines increased the infiltration of activated effector T cells and inhibited tumour growth [305]. Two related clinical trials (Clinicaltrials.gov Identifier: NCT03730948, NCT02919644) on DC-based vaccines are ongoing to develop strategies for preventing the progression of surgically resected stage I and II hypermutated CRC or curatively resected stage IV CRC. Moreover, peptide-based vaccines have strong specificity and can easily elicit an effective immune response [306, 307]. In a phase I clinical trial, the adenovirus (Ad5)-GUCY2C-PADRE vaccine was efficient in patients with CRC and did not cause grade-3/4 toxic events during the 6-month follow-up period after vaccination [308]. Another phase I study demonstrated that the combination of a single-dose PolyPEPI1018 vaccine and maintenance therapy with fluoropyrimidine and bevacizumab was strongly effective, with 96% of vaccine peptides inducing T-cell responses without causing grade 3 or higher adverse events in patients with mCRC [309].
Based on these encouraging results, cancer vaccines may represent an immunotherapeutic strategy that is not limited by the DNA MSI or MMR status. Cancer vaccines can stimulate immune surveillance to combat initially undetected microscopic lesions and consequently enhance the survival of patients with CRLM.
There are multiple therapeutic options for CRLM; however, they fail to meet the requirements for a disease-free prognosis (Fig. 5). Immunotherapy is an emerging and effective weapon in the fight against CRLM that requires further research to explore its superior potential value.

Therapies for CRLM
Discussion and perspectives
Liver metastasis is the most common site of metastasis in CRC and is the leading cause of death in patients with CRC. The liver is a characteristic immune-tolerant organ in which resident liver cells, recruited inflammatory and immune cells and active protein molecules interact with each other; additionally, EVs act as important mediators of intercellular communication. The intricate characteristics and mechanisms of the hepatic immune microenvironment that directly or indirectly induce immunosuppression and contribute to the regulation of cancer metastasis should be extensively investigated to explore potential therapeutic targets for CRLM.
The formation of a PMN in CRC is an important prerequisite for LM, which is a progressive process that triggers local changes such as vascular leakage, ECM remodelling and systemic effects on the immune system. Induced by the combined systemic effects of CRC-secreted factors and EVs, the PMN shapes a microenvironment that is favourable for LM, which makes the distant liver a favourable site for the colonisation of disseminated cancer cells. However, the underlying mechanisms responsible for the formation of the PMN remain uncertain and warrant further investigations.
ICB-based treatment is only effective in patients with MSI-H/dMMR CRC but not in patients with MSS/pMMR CRC. Moreover, another challenge involves the potentially deleterious side effect known as hyperprogression that occurs in some patients after ICB therapy and is independently associated with advanced age and higher metastatic load. With the increasing use of ICB therapy in clinical practice, more studies are required to elucidate the potential mechanisms and to identify the predictors of hyperprogression, which would allow for patients at high risk for life-threatening immune-related adverse events to be screened before ICB therapy. As emerging immunotherapeutic strategies, CAR-T-cell therapy and cancer vaccines may revolutionise the era of cancer immunotherapy. Despite the excellent efficacy of CAR-T-cell therapy in haematological malignancies, its use in solid tumours may be limited due to the trafficking barriers of CAR-T cells, their weak ability to infiltrate solid tumours and their off-target effects. Furthermore, tumour vaccines are a type of individualised immunotherapy with high specificity and few side effects. However, the effective translation of cancer vaccines in clinical practice remains challenging. Further preclinical and clinical trials should be conducted to demonstrate the efficacy of immunotherapy in mCRC and to identify novel therapeutic targets or to develop combination strategies to improve or activate antitumour immune responses for the effective treatment of CRLM.
Availability of data and materials
Not applicable.
Abbreviations
- CRC:
-
Colorectal cancer
- LM:
-
Liver metastasis
- CRLM:
-
Colorectal liver metastasis
- TME:
-
Tumor microenvironment
- ECM:
-
Extracellular matrix
- EV:
-
Extracellular vesicle
- Treg :
-
Regulatory T cell
- MDSC:
-
Myeloid-derived suppressor cell
- TAM:
-
Tumor associated macrophage
- APC:
-
Antigen-presenting cell
- LSEC:
-
Liver sinusoidal endothelial cell
- KC:
-
Kupffer cell
- HSC:
-
Hepatic stellate cell
- DC:
-
Dendritic cell
- NK:
-
Natural killer cell
- MHC:
-
Major histocompatibility complex
- PD-L1:
-
Programmed cell death 1 ligand 1
- PD-1:
-
Programmed cell death-1
- PRF:
-
Perforin
- GZM:
-
Granzyme
- IFNγ:
-
Interferon-γ
- CEA:
-
Carcinoembryonic antigen
- mCRC:
-
Metastatic CRC
- TNF-α:
-
Tumor necrosis factor-α
- IL-1α:
-
Interleukin-1α
- VEGF:
-
Vascular endothelial growth factor
- MMP:
-
Matrix metalloproteinase
- CTLA-4:
-
Cytotoxic T lymphocyte antigen-4
- A2AR:
-
A2A receptor
- EMT:
-
Epithelial-mesenchymal transition
- CTL:
-
Cytotoxic T lymphocyte
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- PMN-MDSCs:
-
Polymorphonuclear MDSCs
- M-MDSCs:
-
Monocytic MDSCs
- S1PR1:
-
Sphingosine-1-phosphate receptor 1
- cNK:
-
Conventional NK cell
- LrNK:
-
Liver-resident natural killer cell
- cDC:
-
Conventional DC
- pDC:
-
Plasmacytoid DC
- TAN:
-
Tumor-associated neutrophil
- NET:
-
Neutrophil extracellular trap
- EGFR:
-
Epithelial growth factor receptor
- PFS:
-
Progression-free survival
- OS:
-
Overall survival
- ICB:
-
Immune checkpoint blockade
- MSI:
-
Microsatellite instability
- MSS:
-
Microsatellite stable
- MMR:
-
Mismatch repair
- ORR:
-
Objective response rate
- CAR-T:
-
Chimeric antigen receptor T cell
- GUCY2C:
-
Guanylyl cyclase C
- NKG2D:
-
Natural killer group 2D
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–49.
Zheng R, Zhang S, Zeng H, Wang S, Sun K, Chen R, et al. Cancer incidence and mortality in China, 2016. Journal of the National Cancer Center. 2022 [cited 2022 Apr 1]; Available from: https://www.sciencedirect.com/science/article/pii/S2667005422000047.
Buccafusca G, Proserpio I, Tralongo AC, Rametta Giuliano S, Tralongo P. Early colorectal cancer: diagnosis, treatment and survivorship care. Critical Reviews in Oncology/Hematology. 2019 ;136:20–30. Available from: https://www.sciencedirect.com/science/article/pii/S1040842817304511. [cited 2022 Aug 4].
Ganesh K, Stadler ZK, Cercek A, Mendelsohn RB, Shia J, Segal NH, et al. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nat Rev Gastroenterol Hepatol. 2019;16:361–75.
Li J, Yuan Y, Yang F, Wang Y, Zhu X, Wang Z, et al. Expert consensus on multidisciplinary therapy of colorectal cancer with lung metastases (2019 edition). Journal of Hematology & Oncology. 2019;12:16 Available from: https://doi.org/10.1186/s13045-019-0702-0. [cited 2022 Aug 4].
Valderrama-Treviño AI, Barrera-Mera B, Ceballos-Villalva JC, Montalvo-Javé EE. Hepatic Metastasis from Colorectal Cancer. Euroasian J Hepatogastroenterol. 2017;7:166–75.https://www.sciencedirect.com/science/article/pii/S2667005422000047.
Fumagalli A, Suijkerbuijk SJE, Begthel H, Beerling E, Oost KC, Snippert HJ, et al. A surgical orthotopic organoid transplantation approach in mice to visualize and study colorectal cancer progression. Nat Protoc Nature Publishing Group. 2018;13:235–47. Available from: https://www.nature.com/articles/nprot.2017.137. [cited 2022 Feb 12].
van der Pool AEM, Damhuis RA, Ijzermans JNM, de Wilt JHW, Eggermont AMM, Kranse R, et al. Trends in incidence, treatment and survival of patients with stage IV colorectal cancer: a population-based series. Colorectal Dis. 2012;14:56–61.
van der Geest LGM, Lam-Boer J, Koopman M, Verhoef C, Elferink MAG, de Wilt JHW. Nationwide trends in incidence, treatment and survival of colorectal cancer patients with synchronous metastases. Clin Exp Metastasis. 2015;32:457–65. Available from: https://doi.org/10.1007/s10585-015-9719-0. [cited 2022 Apr 1].
Wang H, Li X, Peng R, Wang Y, Wang J. Stereotactic ablative radiotherapy for colorectal cancer liver metastasis. Seminars in Cancer Biology. 2021;71:21–32. Available from: https://www.sciencedirect.com/science/article/pii/S1044579X20301541. [cited 2022 Apr 1].
Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA: A Cancer Journal for Clinicians. 2017;67:177–93. Available from: https://onlinelibrary.wiley.com/doi/abs/10.3322/caac.21395. [cited 2022 Feb 11].
Biller LH, Schrag D. Diagnosis and Treatment of Metastatic Colorectal Cancer: A Review. JAMA. 2021;325:669–85.
Capdevila J, Saura C, Macarulla T, Casado E, Ramos FJ, Tabernero J. Monoclonal antibodies in the treatment of advanced colorectal cancer. Eur J Surg Oncol. 2007;33:S24-34. Available from: https://www.sciencedirect.com/science/article/pii/S0748798307005446. [cited 2022 Feb 11].
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Letters. 2017;387:61–8. Available from: https://www.sciencedirect.com/science/article/pii/S0304383516300155. [cited 2022 May 6].
**ao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacology & Therapeutics. 2021;221:107753. Available from: https://www.sciencedirect.com/science/article/pii/S0163725820302849. [cited 2022 May 6].
Wculek SK, Malanchi I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature. 2015;528:413–7.
Hanna RN, Cekic C, Sag D, Tacke R, Thomas GD, Nowyhed H, et al. Patrolling monocytes control tumor metastasis to the lung. Science. 2015;350:985–90.
Nakamura K, Smyth MJ. Myeloid immunosuppression and immune checkpoints in the tumor microenvironment. Cell Mol Immunol Nature Publishing Group. 2020;17:1–12. Available from: https://www.nature.com/articles/s41423-019-0306-1. [cited 2022 May 6].
Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Science. 2019;110:2080–9. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/cas.14069. [cited 2022 Mar 26].
De Cicco P, Ercolano G, Ianaro A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Frontiers in Immunology. 2020;11. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2020.01680. [cited 2022 May 15].
Dou L, Ono Y, Chen Y, Thomson AW, Chen X. Hepatic Dendritic Cells, the Tolerogenic Liver Environment, and Liver Disease. Semin Liver Dis Thieme Medical Publishers. 2018;38:170–80. Available from: http://www.thieme-connect.de/DOI/DOI?10.1055/s-0038-1646949. [cited 2022 Feb 11].
Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol Nature Publishing Group. 2010;10:753–66. Available from: https://www.nature.com/articles/nri2858. [cited 2022 Feb 13].
Heymann F, Peusquens J, Ludwig-Portugall I, Kohlhepp M, Ergen C, Niemietz P, et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology. 2015;62:279–91. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.27793. [cited 2022 Feb 13].
Zhou S-N, Pan W-T, Pan M-X, Luo Q-Y, Zhang L, Lin J-Z, et al. Comparison of Immune Microenvironment Between Colon and Liver Metastatic Tissue in Colon Cancer Patients with Liver Metastasis. Dig Dis Sci. 2021;66:474–82.
Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med. 2021;27:152-64.
Yuzhalin AE, Yu D. Brain Metastasis Organotropism. Cold Spring Harb Perspect Med. 2020;10:a037242.
Weidle UH, Birzele F, Kollmorgen G, Rüger R. Molecular Basis of Lung Tropism of Metastasis. Cancer Genomics Proteomics. 2016;13:129–39.
Minn AJ, Kang Y, Serganova I, Gupta GP, Giri DD, Doubrovin M, et al. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J Clin Invest. 2005;115:44–55. Available from: https://www.jci.org/articles/view/22320#B3. [cited 2022 Feb 13].
Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 Promotes Osteolytic Expansion of Indolent Bone Micrometastasis of Breast Cancer by Engaging α4β1-Positive Osteoclast Progenitors. Cancer Cell. 2011;20:701–14. Available from: https://www.sciencedirect.com/science/article/pii/S1535610811004089. [cited 2022 Feb 13].
Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature Nature Publishing Group. 2011;475:222–5. Available from: https://www.nature.com/articles/nature10138. [cited 2022 Feb 13].
Sevenich L, Bowman RL, Mason SD, Quail DF, Rapaport F, Elie BT, et al. Analysis of tumour- and stroma-supplied proteolytic networks reveals a brain-metastasis-promoting role for cathepsin S. Nat Cell Biol Nature Publishing Group. 2014;16:876–88. Available from: https://www.nature.com/articles/ncb3011. [cited 2022 Feb 13].
Kitamura T, Qian B-Z, Soong D, Cassetta L, Noy R, Sugano G, et al. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J Exp Med. 2015;212:1043–59. https://doi.org/10.1084/jem.20141836. [cited 2022 Feb 13].
Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol Nature Publishing Group. 2016;18:549–60. Available from: https://www.nature.com/articles/ncb3340. [cited 2022 Feb 13].
Roe J-S, Hwang C-I, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017;170:875-888.e20. Available from: https://www.sciencedirect.com/science/article/pii/S0092867417308140. [cited 2022 Feb 13].
Zhao S, Mi Y, Guan B, Zheng B, Wei P, Gu Y, et al. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J Hematol Oncol. 2020;13:156. Available from: https://doi.org/10.1186/s13045-020-00991-2. [cited 2022 Feb 13].
Zhao S, Guan B, Mi Y, Shi D, Wei P, Gu Y, et al. LncRNA MIR17HG promotes colorectal cancer liver metastasis by mediating a glycolysis-associated positive feedback circuit. Oncogene. 2021;40:4709–24.
Budczies J, von Winterfeld M, Klauschen F, Bockmayr M, Lennerz JK, Denkert C, et al. The landscape of metastatic progression patterns across major human cancers. Oncotarget. 2015;6:570–83.
Hess KR, Varadhachary GR, Taylor SH, Wei W, Raber MN, Lenzi R, et al. Metastatic patterns in adenocarcinoma. Cancer. 2006;106:1624–33. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cncr.21778. [cited 2022 Feb 11].
Chow FC-L, Chok KS-H. Colorectal liver metastases: An update on multidisciplinary approach World J Hepatol. 2019;11:150-72.
Mielgo A, Schmid MC. Liver Tropism in Cancer: The Hepatic Metastatic Niche. Cold Spring Harb Perspect Med Cold Spring Harbor Laboratory Press. 2020;10:a037259. Available from: http://perspectivesinmedicine.cshlp.org/content/10/3/a037259. [cited 2022 Feb 13].
Bosch FX, Ribes J, Díaz M, Cléries R. Primary liver cancer: worldwide incidence and trends. Gastroenterology. 2004;127:S5-16.
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer Nature Publishing Group. 2002;2:563–72. Available from: https://www.nature.com/articles/nrc865. [cited 2022 Aug 5].
Lake-Bakaar G, Ahmed M, Evenson A, Bonder A, Faintuch S, Sundaram V. Management of Hepatocellular Carcinoma in Cirrhotic Patients with Portal Hypertension: Relevance of Hagen-Poiseuille’s Law. Liver Cancer. 2014;3:428–38.
Poisson J, Lemoinne S, Boulanger C, Durand F, Moreau R, Valla D, et al. Liver sinusoidal endothelial cells: Physiology and role in liver diseases. J Hepatol. 2017;66:212–27.
Heymann F, Tacke F. Immunology in the liver — from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13:88–110. Available from: https://www.nature.com/articles/nrgastro.2015.200. [cited 2022 Aug 6].
Zheng M, Tian Z. Liver-Mediated Adaptive Immune Tolerance. Front Immunol. 2019;10:2525.
Milette S, Sicklick JK, Lowy AM, Brodt P. Molecular Pathways: Targeting the Microenvironment of Liver Metastases. Clinical Cancer Res. 2017;23:6390–9. Available from: https://doi.org/10.1158/1078-0432.CCR-15-1636. [cited 2022 Aug 6].
Wang X, Huang S, Lu X, Huang Y, Chi P. Incidence of and Risk Factors for Gastroepiploic Lymph Node Involvement in Patients with Cancer of the Transverse Colon Including the Hepatic Flexure. World J Surg. 2021;45:1514–25. Available from: https://doi.org/10.1007/s00268-020-05933-0. [cited 2022 Aug 6].
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
Kim J, Takeuchi H, Lam ST, Turner RR, Wang H-J, Kuo C, et al. Chemokine receptor CXCR4 expression in colorectal cancer patients increases the risk for recurrence and for poor survival. J Clin Oncol. 2005;23:2744–53.
Ghadjar P, Coupland SE, Na I-K, Noutsias M, Letsch A, Stroux A, et al. Chemokine Receptor CCR6 Expression Level and Liver Metastases in Colorectal Cancer. JCO Wolters Kluwer. 2006;24:1910–6. Available from: https://ascopubs.org/doi/10.1200/JCO.2005.04.1822. [cited 2022 Aug 6].
Giakoustidis A, Mudan S, Hagemann T. Tumour Microenvironment: Overview with an Emphasis on the Colorectal Liver Metastasis Pathway. Cancer Microenviron. 2015;8:177–86.
Brodt P. Role of the Microenvironment in Liver Metastasis: From Pre- to Prometastatic Niches. Clin Cancer Res. 2016;22:5971–82 (American Association for Cancer Research). Available from: https://clincancerres.aacrjournals.org/content/22/24/5971. [cited 2022 Feb 15].
Timmers M, Vekemans K, Vermijlen D, Asosingh K, Kuppen P, Bouwens L, et al. Interactions between rat colon carcinoma cells and Kupffer cells during the onset of hepatic metastasis. Int J Cancer. 2004;112:793–802. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/ijc.20481. [cited 2022 Mar 21].
Piñeiro Fernández J, Luddy KA, Harmon C, O’Farrelly C. Hepatic Tumor Microenvironments and Effects on NK Cell Phenotype and Function. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2019;20:4131. Available from: https://www.mdpi.com/1422-0067/20/17/4131. [cited 2022 Aug 6].
Liu X, Xu J, Rosenthal S, Zhang L, McCubbin R, Meshgin N, et al. Identification of Lineage-Specific Transcription Factors That Prevent Activation of Hepatic Stellate Cells and Promote Fibrosis Resolution. Gastroenterology. 2020;158:1728-1744.e14. Available from: https://www.sciencedirect.com/science/article/pii/S0016508520301177. [cited 2022 Feb 13].
Lee J, Ung A, Kim H, Lee K, Cho H-J, Bandaru P, et al. Engineering liver microtissues to study the fusion of HepG2 with mesenchymal stem cells and invasive potential of fused cells. Biofabrication. 2021;14.
Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol. 2007;171:1608–18.
Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–38.
Trefts E, Gannon M, Wasserman DH. The liver. Current Biology. 2017;27:R1147-51. Available from: https://www.sciencedirect.com/science/article/pii/S0960982217311831. [cited 2022 Feb 11].
Hazari Y, Bravo-San Pedro JM, Hetz C, Galluzzi L, Kroemer G. Autophagy in hepatic adaptation to stress. J Hepatol. 2020;72:183–96. Available from: https://www.sciencedirect.com/science/article/pii/S0960982217311831. [cited 2022 Feb 11].
Mikulak J, Bruni E, Oriolo F, Di Vito C, Mavilio D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Frontiers in Immunology. 2019;10. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2019.00946. [cited 2022 Feb 11].
Gracia-Sancho J, Caparrós E, Fernández-Iglesias A, Francés R. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18:411–31.
Abumrad NA, Cabodevilla AG, Samovski D, Pietka T, Basu D, Goldberg IJ. Endothelial Cell Receptors in Tissue Lipid Uptake and Metabolism. Circ Res. 2021;128:433–50.
Tran S, Baba I, Poupel L, Dussaud S, Moreau M, Gélineau A, et al. Impaired Kupffer Cell Self-Renewal Alters the Liver Response to Lipid Overload during Non-alcoholic Steatohepatitis. Immunity. 2020;53:627-640.e5.
Dai S, Liu F, Qin Z, Zhang J, Chen J, Ding W-X, et al. Kupffer cells promote T-cell hepatitis by producing CXCL10 and limiting liver sinusoidal endothelial cell permeability. Theranostics. 2020;10:7163–77.
Trivedi P, Wang S, Friedman SL. The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021;33:242–57.
Hall Z, Chiarugi D, Charidemou E, Leslie J, Scott E, Pellegrinet L, et al. Lipid Remodeling in Hepatocyte Proliferation and Hepatocellular Carcinoma. Hepatology. 2021;73:1028–44.
Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63.
Hudspeth K, Pontarini E, Tentorio P, Cimino M, Donadon M, Torzilli G, et al. The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun. 2013;46:55–65.
Knolle PA, Wohlleber D. Immunological functions of liver sinusoidal endothelial cells. Cell Mol Immunol. 2016;13:347–53.
Pandey E, Nour AS, Harris EN. Prominent Receptors of Liver Sinusoidal Endothelial Cells in Liver Homeostasis and Disease. Front Physiol. 2020;11:873.
Szafranska K, Kruse LD, Holte CF, McCourt P, Zapotoczny B. The wHole Story About Fenestrations in LSEC. Front Physiol. 2021;12:735573.
Géraud C, Evdokimov K, Straub BK, Peitsch WK, Demory A, Dörflinger Y, et al. Unique Cell Type-Specific Junctional Complexes in Vascular Endothelium of Human and Rat Liver Sinusoids. PLOS ONE. Public Library of Science; 2012;7:e34206. Available from: https://journals.plos.org/plosone/article?id=https://doi.org/10.1371/journal.pone.0034206. [cited 2022 Feb 12].
Maslak E, Gregorius A, Chlopicki S. Liver sinusoidal endothelial cells (LSECs) function and NAFLD; NO-based therapy targeted to the liver. Pharmacol Rep. 2015;67:689–94.
Mönkemöller V, Øie C, Hübner W, Huser T, McCourt P. Multimodal super-resolution optical microscopy visualizes the close connection between membrane and the cytoskeleton in liver sinusoidal endothelial cell fenestrations. Sci Rep. 2015;5:16279. Available from: https://www.nature.com/articles/srep16279. [cited 2022 Feb 12].
Smedsrød B, Le Couteur D, Ikejima K, Jaeschke H, Kawada N, Naito M, et al. Hepatic sinusoidal cells in health and disease: update from the 14th International Symposium. Liver Int. 2009;29:490–501. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1478-3231.2009.01979.x. [cited 2022 Feb 12].
Sørensen KK, McCourt P, Berg T, Crossley C, Couteur DL, Wake K, et al. The scavenger endothelial cell: a new player in homeostasis and immunity. Am J Physiol Regul Integr Comp Physiol. 2012;303:R1217-30. Available from: https://journals.physiology.org/doi/full/10.1152/ajpregu.00686.2011. [cited 2022 Feb 12].
Shetty S, Lalor PF, Adams DH. Liver sinusoidal endothelial cells - gatekeepers of hepatic immunity. Nat Rev Gastroenterol Hepatol. 2018;15:555–67.
Diehl L, Schurich A, Grochtmann R, Hegenbarth S, Chen L, Knolle PA. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology. 2008;47:296–305. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.21965. [cited 2022 May 6].
Braet F, Nagatsuma K, Saito M, Soon L, Wisse E, Matsuura T. The hepatic sinusoidal endothelial lining and colorectal liver metastases. World Journal of Gastroenterology. Baishideng Publishing Group Inc.; 2007;13:821–5. Available from: https://www.wjgnet.com/1007-9327/full/v13/i6/821.htm. [cited 2022 Mar 21].
Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome–mediated bile acid metabolism regulates liver cancer via NKT cells. Science. American Association for the Advancement of Science; 2018;360:eaan5931. Available from: https://www.science.org/doi/10.1126/science.aan5931. [cited 2022 Jul 30].
Brodt P, Fallavollita L, Bresalier RS, Meterissian S, Norton CR, Wolitzky BA. Liver endothelial E-selectin mediates carcinoma cell adhesion and promotes liver metastasis. Int J Cancer. 1997;71:612–9. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/%28SICI%291097-0215%2819970516%2971%3A4%3C612%3A%3AAID-IJC17%3E3.0.CO%3B2-D. [cited 2022 May 6].
Bird NC, Mangnall D, Majeed AW. Biology of colorectal liver metastases: A review. J Surg Oncol. 2006;94:68–80. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jso.20558. [cited 2022 May 6].
Brodt P. Role of the Host Inflammatory Response in Colon Carcinoma Initiation. Dordrecht: Progression and Liver Metastasis. Metastasis of Colorectal Cancer. Springer; 2010. p. 289–319.https://linkspringer.53yu.com/chapter/10.1007/978-90-481-8833-8_10. [cited 2022 May 6].
Van den Eynden GG, Majeed AW, Illemann M, Vermeulen PB, Bird NC, Høyer-Hansen G, et al. The Multifaceted Role of the Microenvironment in Liver Metastasis: Biology and Clinical Implications. Cancer Res. 2013;73:2031–43. Available from: https://doi.org/10.1158/0008-5472.CAN-12-3931. [cited 2022 Mar 29].
Liu W, Tang L, Zhang G, Wei H, Cui Y, Guo L, et al. Characterization of a novel C-type lectin-like gene, LSECtin: demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J Biol Chem. 2004;279:18748–58.
Tang L, Yang J, Liu W, Tang X, Chen J, Zhao D, et al. Liver sinusoidal endothelial cell lectin, LSECtin, negatively regulates hepatic T-cell immune response. Gastroenterology. 2009;137:1498-1508 e1 5.
Zuo Y, Ren S, Wang M, Liu B, Yang J, Kuai X, et al. Novel roles of liver sinusoidal endothelial cell lectin in colon carcinoma cell adhesion, migration and in-vivo metastasis to the liver. Gut BMJ Publishing Group. 2013;62:1169–78. Available from: https://gut.bmj.com/content/62/8/1169. [cited 2022 Mar 29].
Yu X, Chen L, Liu J, Dai B, Xu G, Shen G, et al. Immune modulation of liver sinusoidal endothelial cells by melittin nanoparticles suppresses liver metastasis. Nat Commun. 2019;10:574. Available from: http://www.nature.com/articles/s41467-019-08538-x. [cited 2021 Oct 28].
Cai J, Zhang X-J, Li H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology. 2019;70:1026–37. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.30506. [cited 2022 Feb 13].
Shi J, Fujieda H, Kokubo Y, Wake K. Apoptosis of neutrophils and their elimination by Kupffer cells in rat liver. Hepatology. 1996;24:1256–63.
Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer Cells in the Liver. Comprehensive Physiology. John Wiley & Sons: Ltd; 2013. p. 785–97. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/cphy.c120026. [cited 2022 Feb 13].
Khatib A-M, Auguste P, Fallavollita L, Wang N, Samani A, Kontogiannea M, et al. Characterization of the Host Proinflammatory Response to Tumor Cells during the Initial Stages of Liver Metastasis. Am J Pathol. 2005;167:749–59. Available from: https://www.sciencedirect.com/science/article/pii/S0002944010620482. [cited 2022 Mar 21].
Matsumura H, Kondo T, Ogawa K, Tamura T, Fukunaga K, Murata S, et al. Kupffer cells decrease metastasis of colon cancer cells to the liver in the early stage. Int J Oncol. 2014;45:2303–10. Available from: https://www.spandidos-publications.com/10.3892/ijo.2014.2662. [cited 2022 May 7].
Kimura Y, Inoue A, Hangai S, Saijo S, Negishi H, Nishio J, et al. The innate immune receptor Dectin-2 mediates the phagocytosis of cancer cells by Kupffer cells for the suppression of liver metastasis. Proceedings of the National Academy of Sciences of the United States of America National Academy of Sciences; 2016;113:14097. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5150405/. [cited 2023 May 3].
Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. Role of the Kupffer Cell in Mediating Hepatic Toxicity and Carcinogenesis. Toxicol Sci. 2007;96:2–15. Available from: https://doi.org/10.1093/toxsci/kfl173. [cited 2022 Feb 13].
Keirsse J, Van Damme H, Geeraerts X, Beschin A, Raes G, Van Ginderachter JA. The role of hepatic macrophages in liver metastasis. Cell Immunol. 2018;330:202–15.Available from: https://www.sciencedirect.com/science/article/pii/S0008874918301424. [cited 2022 Mar 30].
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–26. Available from: https://www.nature.com/articles/ncb3169. [cited 2023 May 4].
Jiang K, Chen H, Fang Y, Chen L, Zhong C, Bu T, et al. Exosomal ANGPTL1 attenuates colorectal cancer liver metastasis by regulating Kupffer cell secretion pattern and impeding MMP9 induced vascular leakiness. J Exp Clin Cancer Res. 2021;40:21. Available from: https://doi.org/10.1186/s13046-020-01816-3. [cited 2022 Feb 11].
Sun H, Meng Q, Shi C, Yang H, Li X, Wu S, et al. Hypoxia-Inducible Exosomes Facilitate Liver-Tropic Premetastatic Niche in Colorectal Cancer. Hepatology. 2021;74:2633–51.
Liu W, Zhou X, Yao Q, Chen C, Zhang Q, Ding K, et al. In situ expansion and reprogramming of Kupffer cells elicit potent tumoricidal immunity against liver metastasis. J Clin Invest. American Society for Clinical Investigation; 2023;133. Available from: https://webvpn.fudan.edu.cn/https/77726476706e69737468656265737421e7e056d22d33611e711a8e/articles/view/157937. [cited 2023 May 4].
Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–72.
Puche JE, Saiman Y, Friedman SL. Hepatic stellate cells and liver fibrosis. Compr Physiol. 2013;3:1473–92. Available from: http://www.thieme-connect.de/DOI/DOI?10.1055/s-0040-1708876. [cited 2023 Feb 2].
Matsuda M, Seki E. Hepatic Stellate Cell-Macrophage Crosstalk in Liver Fibrosis and Carcinogenesis. Semin Liver Dis. 2020;40:307–20.
Yin C, Evason KJ, Asahina K, Stainier DYR. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–10.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–43. Available from: http://www.nature.com/articles/nature25492. [cited 2021 Nov 26].
Itatani Y, Kawada K, Sakai Y. Transforming Growth Factor-β Signaling Pathway in Colorectal Cancer and Its Tumor Microenvironment. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2019;20:5822. Available from: https://www.mdpi.com/1422-0067/20/23/5822. [cited 2022 Aug 7].
Coulouarn C. Modulating the activation of hepatic stellate cells: A cunning way for metastatic cells to create a permissive soil for seeding in the liver? Hepatology. 2015;61:37–40. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.27330. [cited 2022 Mar 31].
Zhang DY, Goossens N, Guo J, Tsai M-C, Chou H-I, Altunkaynak C, et al. A hepatic stellate cell gene expression signature associated with outcomes in hepatitis C cirrhosis and hepatocellular carcinoma after curative resection. Gut. 2016;65:1754–64. Available from: https://gut.bmj.com/content/65/10/1754. [cited 2022 Mar 30].
Affo S, Yu L-X, Schwabe RF. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu Rev Pathol. 2017;12:153–86.
Zhao S, Mi Y, Zheng B, Wei P, Gu Y, Zhang Z, et al. Highly-metastatic colorectal cancer cell released miR-181a-5p-rich extracellular vesicles promote liver metastasis by activating hepatic stellate cells and remodelling the tumour microenvironment. J Extracell Vesicles. 2022;11:e12186. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/jev2.12186. [cited 2022 Mar 30].
Muhanna N, Doron S, Wald O, Horani A, Eid A, Pappo O, et al. Activation of hepatic stellate cells after phagocytosis of lymphocytes: A novel pathway of fibrogenesis. Hepatology. 2008;48:963–77. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.22413. [cited 2022 May 7].
Qi M, Fan S, Huang M, Pan J, Li Y, Miao Q, et al. Targeting FAPα-expressing hepatic stellate cells overcomes resistance to antiangiogenics in colorectal cancer liver metastasis models. J Clin Invest. American Society for Clinical Investigation; 2023;132. Available from: https://www.jci.org/articles/view/157399. [cited 2023 Feb 2].
Damo M, Wilson DS, Watkins EA, Hubbell JA. Soluble N-Acetylgalactosamine-Modified Antigens Enhance Hepatocyte-Dependent Antigen Cross-Presentation and Result in Antigen-Specific CD8+ T Cell Tolerance Development. Frontiers in Immunology. 2021 ;12. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2021.555095. [cited 2022 Jul 31].
Mook ORF, van Marle J, Jonges R, Vreeling-Sindelárová H, Frederiks WM, Van Noorden CJF. Interactions between colon cancer cells and hepatocytes in rats in relation to metastasis. J Cell Mol Med. 2008;12:2052–61.
Shimizu S, Yamada N, Sawada T, Ikeda K, Nakatani K, Seki S, et al. Ultrastructure of early phase hepatic metastasis of human colon carcinoma cells with special reference to desmosomal junctions with hepatocytes. Pathol Int. 2000;50:953–9.
Yoshioka T, Nishikawa Y, Ito R, Kawamata M, Doi Y, Yamamoto Y, et al. Significance of integrin αvβ5 and erbB3 in enhanced cell migration and liver metastasis of colon carcinomas stimulated by hepatocyte-derived heregulin Cancer Science. 2010;101:2011-8. Available from:https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1349-7006.2010.01640.x.[cited 2022 Mar 30].
Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature. 2019;567:249–52.
Wu Y, Brodt P, Sun H, Mejia W, Novosyadlyy R, Nunez N, et al. Insulin-like growth factor-I regulates the liver microenvironment in obese mice and promotes liver metastasis. Cancer Res. 2010;70:57–67.
Wang N, Rayes RF, Elahi SM, Lu Y, Hancock MA, Massie B, et al. The IGF-Trap: Novel Inhibitor of Carcinoma Growth and Metastasis. Molecular Cancer Therapeutics. 2015;14:982–93. Available from: https://doi.org/10.1158/1535-7163.MCT-14-0751. [cited 2022 Mar 30].
Assudani DP, Horton RBV, Mathieu MG, McArdle SEB, Rees RC. The role of CD4+ T cell help in cancer immunity and the formulation of novel cancer vaccines. Cancer Immunol Immunother. 2007;56:70–80. Available from: https://doi.org/10.1007/s00262-006-0154-6. [cited 2022 May 8].
Lee H-G, Cho M-Z, Choi J-M. Bystander CD4+ T cells: crossroads between innate and adaptive immunity. Exp Mol Med. 2020;52:1255–63.
Chalmin F, Mignot G, Bruchard M, Chevriaux A, Végran F, Hichami A, et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity. 2012;36:362–73.
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31:787–98.
Kroemer M, Turco C, Spehner L, Viot J, Idirène I, Bouard A, et al. Investigation of the prognostic value of CD4 T cell subsets expanded from tumor-infiltrating lymphocytes of colorectal cancer liver metastases. J Immunother Cancer. BMJ Specialist Journals; 2020;8:e001478. Available from: https://jitc.bmj.com/content/8/2/e001478. [cited 2022 May 8]
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res. 2011;71:1263–71. Available from: https://doi.org/10.1158/0008-5472.CAN-10-2907. [cited 2022 May 8]
Ji D, Song C, Li Y, **a J, Wu Y, Jia J, et al. Combination of radiotherapy and suppression of Tregs enhances abscopal antitumor effect and inhibits metastasis in rectal cancer. J Immunother Cancer. 2020;8:e000826.
Kumagai S, Koyama S, Itahashi K, Tanegashima T, Lin Y, Togashi Y, et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201-218.e9. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1535610822000034. [cited 2022 Jul 13].
Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer. 2021;9:e002591.
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-Endocytosis of CD80 and CD86: A Molecular Basis for the Cell-Extrinsic Function of CTLA-4. Science. American Association for the Advancement of Science; 2011;332:600-3. Available from: https://www.science.org/doi/10.1126/science.1202947. [cited 2022 May 8].
Walker LSK, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol. 2011;11:852–63. Available from: https://www.nature.com/articles/nri3108. [cited 2022 May 8].
Maj T, Wang W, Crespo J, Zhang H, Wang W, Wei S, et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat Immunol. 2017;18:1332–41.
Setoguchi R, Hori S, Takahashi T, Sakaguchi S. Homeostatic maintenance of natural Foxp3(+) CD25(+) CD4(+) regulatory T cells by interleukin (IL)-2 and induction of autoimmune disease by IL-2 neutralization. J Exp Med. 2005;201:723–35.
Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10:1969–80.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450:566–9.
Katsuno Y, Lamouille S, Derynck R. TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr Opin Oncol. 2013;25:76–84.
Oh E, Hong J, Yun C-O. Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial-Mesenchymal Transition. Cells. 2019;8:E1387.
Farhood B, Najafi M, Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. 2019;234:8509–21.
St. Paul M, Ohashi PS. The Roles of CD8+ T Cell Subsets in Antitumor Immunity. Trends in Cell Biology 2020;30:695-704. Available from: https://www.sciencedirect.com/science/article/pii/S0962892420301215. [cited 2022 Aug 6].
Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature. 2019;571:211–8. Available from: https://www.nature.com/articles/s41586-019-1325-x/. [cited 2022 May 9].
Oshi M, Asaoka M, Tokumaru Y, Yan L, Matsuyama R, Ishikawa T, et al. CD8 T Cell Score as a Prognostic Biomarker for Triple Negative Breast Cancer. Int J Mol Sci. 2020;21:E6968.
Voskoboinik I, Whisstock JC, Trapani JA. Perforin and granzymes: function, dysfunction and human pathology. Nat Rev Immunol. 2015;15:388–400. Available from: https://www.nature.com/articles/nri3839. [cited 2022 Mar 23].
Masuda K, Kornberg A, Miller J, Lin S, Suek N, Botella T, et al. Multiplexed single-cell analysis reveals prognostic and nonprognostic T cell types in human colorectal cancer. JCI Insight. 2022;7:e154646.
Crispe IN. Hepatic T cells and liver tolerance. Nat Rev Immunol. 2003;3:51–62.
Doherty DG. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J Autoimmun. 2016;66:60–75. Available from: https://www.sciencedirect.com/science/article/pii/S089684111530038X. [cited 2023 Apr 30].
Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion.’ Nat Rev Immunol. 2019;19:665–74. Available from: https://www.nature.com/articles/s41577-019-0221-9. [cited 2023 Feb 13].
van der Leun AM, Thommen DS, Schumacher TN. CD8+ T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20:218–32. Available from: https://www.nature.com/articles/s41568-019-0235-4. [cited 2022 Mar 7].
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99.
Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44:1052–68.
Wu Y, Yang S, Ma J, Chen Z, Song G, Rao D, et al. Spatiotemporal Immune Landscape of Colorectal Cancer Liver Metastasis at Single-Cell Level. Cancer Discov. 2022;12:134–53. Available from: https://doi.org/10.1158/2159-8290.CD-21-0316. [cited 2022 Mar 25].
Acharya N, Madi A, Zhang H, Klapholz M, Escobar G, Dulberg S, et al. Endogenous Glucocorticoid Signaling Regulates CD8+ T Cell Differentiation and Development of Dysfunction in the Tumor Microenvironment. Immunity Elsevier. 2020;53:658-671.e6. Available from: https://www.cell.com/immunity/abstract/S1074-7613(20)30358-7. [cited 2022 Aug 2].
Hu Z, Chen G, Zhao Y, Gao H, Li L, Yin Y, et al. Exosome-derived circCCAR1 promotes CD8 + T-cell dysfunction and anti-PD1 resistance in hepatocellular carcinoma. Mol Cancer. 2023;22:55. Available from: https://doi.org/10.1186/s12943-023-01759-1
Rong D, Sun G, Zheng Z, Liu L, Chen X, Wu F, et al. MGP promotes CD8+ T cell exhaustion by activating the NF-κB pathway leading to liver metastasis of colorectal cancer. International Journal of Biological Sciences. Ivyspring International Publisher; 2022;18:2345–61. Available from: https://www.ijbs.com/v18p2345.htm. [cited 2023 Apr 30].
Wu Q, Zhou W, Yin S, Zhou Y, Chen T, Qian J, et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology. 2019;70:198. Available from: https://journals.lww.com/hep/Fulltext/2019/07000/Blocking_Triggering_Receptor_Expressed_on_Myeloid.17.aspx. [cited 2023 May 1]
Cheng L, Du X, Wang Z, Ju J, Jia M, Huang Q, et al. Hyper-IL-15 suppresses metastatic and autochthonous liver cancer by promoting tumour-specific CD8+ T cell responses. J Hepatol. 2014;61:1297–303. Available from: https://www.journal-of-hepatology.eu/article/S0168-8278(14)00472-3/fulltext. [cited 2023 May 1].
Komohara Y, Fujiwara Y, Ohnishi K, Takeya M. Tumor-associated macrophages: Potential therapeutic targets for anti-cancer therapy. Adv Drug Deliv Rev. 2016;99:180–5.
Guerriero JL. Chapter Three - Macrophages: Their Untold Story in T Cell Activation and Function. In: Galluzzi L, Rudqvist N-P, editors. International Review of Cell and Molecular Biology. Academic Press; 2019 . p.73–93. Available from: https://www.sciencedirect.com/science/article/pii/S1937644818300698. [cited 2022 May 10].
Locati M, Curtale G, Mantovani A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu Rev Pathol. 2020;15:123–47. Available from: https://doi.org/10.1146/annurev-pathmechdis-012418-012718. [cited 2023 Feb 8].
Prenen H, Mazzone M. Tumor-associated macrophages: a short compendium. Cell Mol Life Sci. 2019;76:1447–58.
Pan Y, Yu Y, Wang X, Zhang T. Tumor-Associated Macrophages in Tumor Immunity. Frontiers in Immunology. 2020 [cited 2022 Mar 24];11. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2020.583084. [cited 2022 Mar 24].
Huang Y, Snuderl M, Jain RK. Polarization of Tumor-Associated Macrophages: A Novel Strategy for Vascular Normalization and Antitumor Immunity. Cancer Cell. 2011;19:1–2. Available from: https://www.sciencedirect.com/science/article/pii/S1535610811000067. [cited 2022 May 10].
Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: The role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66:1–9. Available from: https://www.sciencedirect.com/science/article/pii/S104084280700162X. [cited 2022 May 10].
Boutilier AJ, Elsawa SF. Macrophage Polarization States in the Tumor Microenvironment. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2021 [cited 2022 May 10];22:6995. Available from: https://www.mdpi.com/1422-0067/22/13/6995. [cited 2022 May 10].
Liu Y, Zhang Q, **ng B, Luo N, Gao R, Yu K, et al. Immune phenotypic linkage between colorectal cancer and liver metastasis. Cancer Cell. Elsevier; 2022 ;0. Available from: https://www.cell.com/cancer-cell/abstract/S1535-6108(22)00065-4. [cited 2022 Mar 18].
Grossman JG, Nywening TM, Belt BA, Panni RZ, Krasnick BA, DeNardo DG, et al. Recruitment of CCR2+ tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. OncoImmunology. Taylor & Francis; 2018;7:e1470729. Available from: https://doi.org/10.1080/2162402X.2018.1470729
Tu W, Gong J, Zhou Z, Tian D, Wang Z. TCF4 enhances hepatic metastasis of colorectal cancer by regulating tumor-associated macrophage via CCL2/CCR2 signaling. Cell Death Dis [Internet]. Nature Publishing Group; 2021 [cited 2023 May 2];12:1–15. Available from: https://www.nature.com/articles/s41419-021-04166-w
Huang C, Ou R, Chen X, Zhang Y, Li J, Liang Y, et al. Tumor cell-derived SPON2 promotes M2-polarized tumor-associated macrophage infiltration and cancer progression by activating PYK2 in CRC. Journal of Experimental & Clinical Cancer Research. 2021 [cited 2022 Oct 9];40:304. Available from: https://doi.org/10.1186/s13046-021-02108-0
Yang P, Qin H, Li Y, **ao A, Zheng E, Zeng H, et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat Commun [Internet]. Nature Publishing Group; 2022 [cited 2023 May 2];13:5782. Available from: https://www.nature.com/articles/s41467-022-33349-y
Wei C, Yang C, Wang S, Shi D, Zhang C, Lin X, et al. Crosstalk between cancer cells and tumor associated macrophages is required for mesenchymal circulating tumor cell-mediated colorectal cancer metastasis. Mol Cancer. 2019;18:64.
Zhang X-L, Hu L-P, Yang Q, Qin W-T, Wang X, Xu C-J, et al. CTHRC1 promotes liver metastasis by resha** infiltrated macrophages through physical interactions with TGF-β receptors in colorectal cancer. Oncogene [Internet]. Nature Publishing Group; 2021 ;40:3959–73. Available from: https://www.nature.com/articles/s41388-021-01827-0. [cited 2023 May 2].
Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell. 2020 ;181:442–459.e29. Available from: https://www.sciencedirect.com/science/article/pii/S009286742030341X. [cited 2023 Feb 8].
Sathe A, Mason K, Grimes SM, Zhou Z, Lau BT, Bai X, et al. Colorectal Cancer Metastases in the Liver Establish Immunosuppressive Spatial Networking between Tumor-Associated SPP1+ Macrophages and Fibroblasts. Clinical Cancer Research [Internet]. 2023 ;29:244–60. Available from: https://doi.org/10.1158/1078-0432.CCR-22-2041. [cited 2023 May 1].
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell. 2014;25:605–20.
Donadon M, Torzilli G, Cortese N, Soldani C, Di Tommaso L, Franceschini B, et al. Macrophage morphology correlates with single-cell diversity and prognosis in colorectal liver metastasis. J Exp Med. 2020;217:20191847. Available from: https://doi.org/10.1084/jem.20191847. [cited 2022 Mar 26].
Liu M, Zhou J, Liu X, Feng Y, Yang W, Wu F, et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut. 2020;69:365–79.
Hoechst B, Ormandy LA, Ballmaier M, Lehner F, Krüger C, Manns MP, et al. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology. 2008 ;135:234–43. Available from: https://www.sciencedirect.com/science/article/pii/S0016508508004563. [cited 2022 Mar 28].
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, et al. Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology. 2009;50:799–807.
Hu C-E, Gan J, Zhang R-D, Cheng Y-R, Huang G-J. Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol. 2011;46:156–64.
Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGFβ Signaling in Mammary Carcinomas Recruits Gr-1+CD11b+ Myeloid Cells that Promote Metastasis. Cancer Cell [Internet]. 2008 ;13:23–35. Available from: https://www.sciencedirect.com/science/article/pii/S153561080700373X. [cited 2022 May 15].
Veglia F, Perego M, Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat Immunol [Internet]. Nature Publishing Group; 2018 ;19:108–19. Available from: https://www.nature.com/articles/s41590-017-0022-x. [cited 2022 May 12].
OuYang L-Y, Wu X-J, Ye S-B, Zhang R, Li Z-L, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med. BioMed Central; 2015 ;13:1–12. Available from: https://translational-medicine.biomedcentral.com/articles/10.1186/s12967-015-0410-7. [cited 2022 May 12].
Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and Tumor-Infiltrating Myeloid-Derived Suppressor Cells in Patients with Colorectal Carcinoma. PLOS ONE . Public Library of Science; 2013 ;8:e57114. Available from: https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0057114. [cited 2022 May 12].
Zeng X, Ward SE, Zhou J, Cheng ASL. Liver Immune Microenvironment and Metastasis from Colorectal Cancer-Pathogenesis and Therapeutic Perspectives. Cancers (Basel). 2021;13:2418.
Wang D, Sun H, Wei J, Cen B, DuBois RN. CXCL1 Is Critical for Premetastatic Niche Formation and Metastasis in Colorectal Cancer. Cancer Res. 2017;77:3655–65.
Itatani Y, Kawada K, Fujishita T, Kakizaki F, Hirai H, Matsumoto T, et al. Loss of SMAD4 from colorectal cancer cells promotes CCL15 expression to recruit CCR1+ myeloid cells and facilitate liver metastasis. Gastroenterology. 2013;145:1064-1075.e11.
Inamoto S, Itatani Y, Yamamoto T, Minamiguchi S, Hirai H, Iwamoto M, et al. Loss of SMAD4 Promotes Colorectal Cancer Progression by Accumulation of Myeloid-Derived Suppressor Cells through the CCL15–CCR1 Chemokine Axis. Clinical Cancer Research. 2016 ;22:492–501. Available from: https://doi.org/10.1158/1078-0432.CCR-15-0726. [cited 2022 Aug 2].
Lin Q, Ren L, Jian M, Xu P, Li J, Zheng P, et al. The mechanism of the premetastatic niche facilitating colorectal cancer liver metastasis generated from myeloid-derived suppressor cells induced by the S1PR1–STAT3 signaling pathway. Cell Death Dis. 2019 ;10:693. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6751205/. [cited 2022 May 12].
Zeng X, Zhou J, **ong Z, Sun H, Yang W, Mok MTS, et al. Cell cycle-related kinase reprograms the liver immune microenvironment to promote cancer metastasis. Cell Mol Immunol [Internet]. Nature Publishing Group; 2021 ;18:1005–15. Available from: https://www.nature.com/articles/s41423-020-00534-2. [cited 2022 Mar 28].
Ren X, **ao J, Zhang W, Wang F, Yan Y, Wu X, et al. Inhibition of CCL7 derived from Mo-MDSCs prevents metastatic progression from latency in colorectal cancer. Cell Death Dis [Internet]. Nature Publishing Group; 2021 ;12:1–15. Available from: https://www.nature.com/articles/s41419-021-03698-5. [cited 2022 May 12].
Prager I, Liesche C, van Ooijen H, Urlaub D, Verron Q, Sandström N, et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J Exp Med. 2019;216:2113–27.
Shimasaki N, Jain A, Campana D. NK cells for cancer immunotherapy. Nat Rev Drug Discov [Internet]. Nature Publishing Group; 2020 ;19:200–18. Available from: https://www.nature.com/articles/s41573-019-0052-1. [cited 2022 Mar 26].
Wu S-Y, Fu T, Jiang Y-Z, Shao Z-M. Natural killer cells in cancer biology and therapy. Mol Cancer. 2020;19:120.
López-Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell. 2017;32:135–54.
Donadon M, Hudspeth K, Cimino M, Di Tommaso L, Preti M, Tentorio P, et al. Increased Infiltration of Natural Killer and T Cells in Colorectal Liver Metastases Improves Patient Overall Survival. J Gastrointest Surg. 2017;21:1226–36.
Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol Res. 2019;7:335–46.
Stiff A, Trikha P, Mundy-Bosse B, McMichael E, Mace TA, Benner B, et al. Nitric Oxide Production by Myeloid-Derived Suppressor Cells Plays a Role in Impairing Fc Receptor-Mediated Natural Killer Cell Function. Clin Cancer Res. 2018;24:1891–904.
Song J, Song H, Wei H, Sun R, Tian Z, Peng H. Requirement of RORα for maintenance and antitumor immunity of liver-resident natural killer cells/ILC1s. Hepatology. [cited 2022 Mar 27];n/a. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.32147. [cited 2022 Mar 27].
Villar J, Segura E. Decoding the Heterogeneity of Human Dendritic Cell Subsets. Trends in Immunology. Elsevier; 2020 ;41:1062–71. Available from: https://www.cell.com/trends/immunology/abstract/S1471-4906(20)30231-3. [cited 2022 Aug 3].
Gerhard GM, Bill R, Messemaker M, Klein AM, Pittet MJ. Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J Exp Med. 2020;218:20200264.
Gupta YH, Khanom A, Acton SE. Control of Dendritic Cell Function Within the Tumour Microenvironment. Front Immunol. 2022;13:733800.
Bošnjak B, Do KTH, Förster R, Hammerschmidt SI. Imaging dendritic cell functions. Immunol Rev. 2022;306:137–63.
Ferris ST, Durai V, Wu R, Theisen DJ, Ward JP, Bern MD, et al. cDC1 prime and are licensed by CD4+ T cells to induce anti-tumour immunity. Nature. 2020;584:624–9.
Balan S, Saxena M, Bhardwaj N. Dendritic cell subsets and locations. Int Rev Cell Mol Biol. 2019;348:1–68.
Duong E, Fessenden TB, Lutz E, Dinter T, Yim L, Blatt S, et al. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity. 2022;55:308-323.e9.
Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity. 2016 ;44:924–38. Available from: https://www.sciencedirect.com/science/article/pii/S1074761316301017. [cited 2022 Mar 27].
Merad M, Sathe P, Helft J, Miller J, Mortha A. The dendritic cell lineage: ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu Rev Immunol. 2013;31:563–604.
Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24.
Ho WW, Gomes-Santos IL, Aoki S, Datta M, Kawaguchi K, Talele NP, et al. Dendritic cell paucity in mismatch repair-proficient colorectal cancer liver metastases limits immune checkpoint blockade efficacy. Proc Natl Acad Sci U S A. 2021;118:e2105323118.
Tokita D, Sumpter TL, Raimondi G, Zahorchak AF, Wang Z, Nakao A, et al. Poor allostimulatory function of liver plasmacytoid DC is associated with pro-apoptotic activity, dependent on regulatory T cells. J Hepatology. 2008;49:1008–18. Available from:https://www.sciencedirect.com/science/article/pii/S016882780800603X. [cited 2022 Mar 27].
Lurje I, Hammerich L, Tacke F. Dendritic Cell and T Cell Crosstalk in Liver Fibrogenesis and Hepatocarcinogenesis: Implications for Prevention and Therapy of Liver Cancer. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2020;21:7378. Available from: https://www.mdpi.com/1422-0067/21/19/7378. [cited 2022 Aug 3]
**a S, Guo Z, Xu X, Yi H, Wang Q, Cao X. Hepatic microenvironment programs hematopoietic progenitor differentiation into regulatory dendritic cells, maintaining liver tolerance. Blood. 2008;112:3175–85. Available from: https://doi.org/10.1182/blood-2008-05-159921. [cited 2022 Mar 27].
**ong S, Dong L, Cheng L. Neutrophils in cancer carcinogenesis and metastasis. J Hematol Oncol. 2021;14:173.
Giese MA, Hind LE, Huttenlocher A. Neutrophil plasticity in the tumor microenvironment. Blood. 2019;133:2159–67. Available from: https://doi.org/10.1182/blood-2018-11-844548. [cited 2022 Aug 4]
Zhou S-L, Zhou Z-J, Hu Z-Q, Huang X-W, Wang Z, Chen E-B, et al. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150:1646-1658.e17.
Zhu K, Li P, Mo Y, Wang J, Jiang X, Ge J, et al. Neutrophils: Accomplices in metastasis. Cancer Letters. 2020;492:11–20. Available from: https://www.sciencedirect.com/science/article/pii/S0304383520303840. [cited 2022 Mar 28].
Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2018;216:176–94. Available from: https://doi.org/10.1084/jem.20181170. [cited 2022 Mar 27].
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil Extracellular Traps Promote the Development and Progression of Liver Metastases after Surgical Stress. Cancer Res. 2016;76:1367–80.
Shang A, Gu C, Zhou C, Yang Y, Chen C, Zeng B, et al. Exosomal KRAS mutation promotes the formation of tumor-associated neutrophil extracellular traps and causes deterioration of colorectal cancer by inducing IL-8 expression. Cell Commun Signal. 2020;18:52.
Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, et al. IL-8 mediates a positive loop connecting increased neutrophil extracellular traps (NETs) and colorectal cancer liver metastasis. J Cancer. 2020;11:4384–96.
Tian S, Chu Y, Hu J, Ding X, Liu Z, Fu D, et al. Tumour-associated neutrophils secrete AGR2 to promote colorectal cancer metastasis via its receptor CD98hc–xCT. Gut . BMJ Publishing Group; 2022 ; Available from: https://gut.bmj.com/content/early/2022/01/26/gutjnl-2021-325137. [cited 2022 Mar 28].
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19:213–28.
Ortiz A. Extracellular vesicles in cancer progression. Seminars in Cancer Biology. 2021;76:139–42. Available from: https://www.sciencedirect.com/science/article/pii/S1044579X21001620. [cited 2022 Aug 6].
Meng W, He C, Hao Y, Wang L, Li L, Zhu G. Prospects and challenges of extracellular vesicle-based drug delivery system: considering cell source. Drug Delivery Taylor & Francis. 2020;27:585–98. Available from: https://doi.org/10.1080/10717544.2020.1748758. [cited 2022 Mar 31].
Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24:766–9.
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523:177–82. Available from: https://www.nature.com/articles/nature14581. [cited 2022 Mar 31]
Shao Y, Chen T, Zheng X, Yang S, Xu K, Chen X, et al. Colorectal cancer-derived small extracellular vesicles establish an inflammatory premetastatic niche in liver metastasis. Carcinogenesis. 2018;39:1368–79. Available from: https://doi.org/10.1093/carcin/bgy115. [cited 2022 Mar 31].
Siveen KS, Raza A, Ahmed EI, Khan AQ, Prabhu KS, Kuttikrishnan S, et al. The Role of Extracellular Vesicles as Modulators of the Tumor Microenvironment, Metastasis and Drug Resistance in Colorectal Cancer. Cancers. Multidisciplinary Digital Publishing Institute; 2019 ;11:746. Available from: https://www.mdpi.com/2072-6694/11/6/746. [cited 2022 Mar 31].
Li B, Cao Y, Sun M, Feng H. Expression, regulation, and function of exosome-derived miRNAs in cancer progression and therapy. The FASEB Journal. 2021;35:e21916. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1096/fj.202100294RR. [cited 2022 Mar 31].
Chang L-C, Chiu H-M, Wu M-S, Shen T-L. The Role of Small Extracellular Vesicles in the Progression of Colorectal Cancer and Its Clinical Applications. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2022;23:1379. Available from: https://www.mdpi.com/1422-0067/23/3/1379
He X, Zhong X, Hu Z, Zhao S, Wei P, Li D. An insight into small extracellular vesicles: Their roles in colorectal cancer progression and potential clinical applications. Clinical and Translational Medicine. 2020 [cited 2021 Dec 2];10. Available from: https://www.mdpi.com/1422-0067/23/3/1379. [cited 2022 Mar 31].
Wei P, Wu F, Kang B, Sun X, Heskia F, Pachot A, et al. Plasma extracellular vesicles detected by Single Molecule array technology as a liquid biopsy for colorectal cancer. J Extracell Vesicles. 2020;9:1809765.
Zeng Z, Li Y, Pan Y, Lan X, Song F, Sun J, et al. Cancer-derived exosomal miR-25–3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat Commun. Nature Publishing Group; 2018;9:5395. Available from: https://www.nature.com/articles/s41467-018-07810-w. [cited 2022 May 15].
Lan J, Sun L, Xu F, Liu L, Hu F, Song D, et al. M2 Macrophage-Derived Exosomes Promote Cell Migration and Invasion in Colon Cancer. Cancer Res. 2019;79:146–58. Available from: https://doi.org/10.1158/0008-5472.CAN-18-0014. [cited 2022 Mar 31].
Zhang C, Wang X-Y, Zhang P, He T-C, Han J-H, Zhang R, et al. Cancer-derived exosomal HSPC111 promotes colorectal cancer liver metastasis by reprogramming lipid metabolism in cancer-associated fibroblasts. Cell Death Dis. Nature Publishing Group; 2022 ;13:1–14. Available from: https://www.nature.com/articles/s41419-022-04506-4. [cited 2022 Mar 31].
Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell. 2016;30:836–48.
A H, B C-S, Tl S, G R, A H, M TM, et al. Tumour exosome integrins determine organotropic metastasis. Nature. Nature; 2015 ;527. Available from: https://pubmed.ncbi.nlm.nih.gov/26524530/. [cited 2022 Aug 4].
Han W, Duan Z. Roles of exosomes in liver metastases: Novel diagnosis and treatment choices. J Cell Physiol. 2019;234:21588–600.
Nordlinger B, Sorbye H, Glimelius B, Poston GJ, Schlag PM, Rougier P, et al. Perioperative FOLFOX4 chemotherapy and surgery versus surgery alone for resectable liver metastases from colorectal cancer (EORTC 40983): long-term results of a randomised, controlled, phase 3 trial. Lancet Oncol. 2013;14:1208–15. Available from: https://www.sciencedirect.com/science/article/pii/S1470204513704479. [cited 2022 Feb 11].
Yin Z, Liu C, Chen Y, Bai Y, Shang C, Yin R, et al. Timing of hepatectomy in resectable synchronous colorectal liver metastases (SCRLM): Simultaneous or delayed? Hepatology. 2013;57:2346–57. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1002/hep.26283. [cited 2022 Apr 1].
Gavriilidis P, Sutcliffe RP, Hodson J, Marudanayagam R, Isaac J, Azoulay D, et al. Simultaneous versus delayed hepatectomy for synchronous colorectal liver metastases: a systematic review and meta-analysis. HPB. 2018;20:11–9. Available from: https://www.sciencedirect.com/science/article/pii/S1365182X1730881X. [cited 2022 Apr 1].
Boudjema K, Locher C, Sabbagh C, Ortega-Deballon P, Heyd B, Bachellier P, et al. Simultaneous Versus Delayed Resection for Initially Resectable Synchronous Colorectal Cancer Liver Metastases: A Prospective, Open-label, Randomized Controlled Trial. Ann Surg. 2021;273:49–56. Available from: https://journals.lww.com/annalsofsurgery/Fulltext/2021/01000/Simultaneous_Versus_Delayed_Resection_for.10.aspx. [cited 2022 Apr 1].
Kanas GP, Taylor A, Primrose JN, Langeberg WJ, Kelsh MA, Mowat FS, et al. Survival after liver resection in metastatic colorectal cancer: review and meta-analysis of prognostic factors. Clin Epidemiol. 2012;4:283–301.
Abdalla EK, Vauthey J-N, Ellis LM, Ellis V, Pollock R, Broglio KR, et al. Recurrence and Outcomes Following Hepatic Resection, Radiofrequency Ablation, and Combined Resection/Ablation for Colorectal Liver Metastases. Ann Surg. 2004;239:818–27. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1356290/. [cited 2022 Apr 2].
Tomasello G, Petrelli F, Ghidini M, Russo A, Passalacqua R, Barni S. FOLFOXIRI Plus Bevacizumab as Conversion Therapy for Patients With Initially Unresectable Metastatic Colorectal Cancer: A Systematic Review and Pooled Analysis. JAMA Oncology. 2017;3:e170278. https://doi.org/10.1001/jamaoncol.2017.0278.
Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen Y-J, Ciombor KK, et al. Colon Cancer, Version 2.2021, NCCN Clinical Practice Guidelines in Oncology. Journal of the National Comprehensive Cancer Network. National Comprehensive Cancer Network; 2021;19:329–59. Available from: https://jnccn.org/view/journals/jnccn/19/3/article-p329.xml. [cited 2022 Apr 3].
Adam R, Delvart V, Pascal G, Valeanu A, Castaing D, Azoulay D, et al. Rescue surgery for unresectable colorectal liver metastases downstaged by chemotherapy: a model to predict long-term survival. Ann Surg. 2004;240:644–57 (discussion 657-658).
Li F, Zhao C, Wang L. Molecular-targeted agents combination therapy for cancer: developments and potentials. Int J Cancer. 2014;134:1257–69.
Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15:569–79.
**e Y-H, Chen Y-X, Fang J-Y. Comprehensive review of targeted therapy for colorectal cancer. Sig Transduct Target Ther. Nature Publishing Group; 2020 [cited 2022 Apr 4];5:1–30. Available from: https://www.nature.com/articles/s41392-020-0116-z. [cited 2022 Apr 4].
Cunningham D, Lang I, Marcuello E, Lorusso V, Ocvirk J, Shin DB, et al. Bevacizumab plus capecitabine versus capecitabine alone in elderly patients with previously untreated metastatic colorectal cancer (AVEX): an open-label, randomised phase 3 trial. Lancet Oncol. 2013;14:1077–85. Available from: https://www.sciencedirect.com/science/article/pii/S1470204513701542. [cited 2022 Apr 4].
Hurwitz HI, Tebbutt NC, Kabbinavar F, Giantonio BJ, Guan Z-Z, Mitchell L, et al. Efficacy and safety of bevacizumab in metastatic colorectal cancer: pooled analysis from seven randomized controlled trials. Oncologist. 2013;18:1004–12.
Baraniskin A, Buchberger B, Pox C, Graeven U, Holch JW, Schmiegel W, et al. Efficacy of bevacizumab in first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Eur J Cancer. 2019;106:37–44.
Abdalla AME, **ao L, Ullah MW, Yu M, Ouyang C, Yang G. Current Challenges of Cancer Anti-angiogenic Therapy and the Promise of Nanotherapeutics. Theranostics. 2018;8:533–48.
Lugano R, Ramachandran M, Dimberg A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol Life Sci. 2020;77:1745–70. https://doi.org/10.1007/s00018-019-03351-7. [cited 2022 May 7].
Hoos A, Ibrahim R, Korman A, Abdallah K, Berman D, Shahabi V, et al. Development of Ipilimumab: Contribution to a New Paradigm for Cancer Immunotherapy. Semin Oncol. 2010;37:533–46.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N Engl J Med. Massachusetts Medical Society; 2010;363:711–23. Available from: https://doi.org/10.1056/NEJMoa1003466. [cited 2022 Apr 5].
Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. Massachusetts Medical Society; 2015;372:2521-32. Available from: https://doi.org/10.1056/NEJMoa1503093. [cited 2022 Apr 5].
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015;373:1803–13.
Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:1627–39.
Herbst RS, Baas P, Kim D-W, Felip E, Pérez-Gracia JL, Han J-Y, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. The Lancet. 2016 ;387:1540–50. Available from: https://www.sciencedirect.com/science/article/pii/S0140673615012817. [cited 2022 Apr 5].
Johdi NA, Sukor NF. Colorectal Cancer Immunotherapy: Options and Strategies. Frontiers in Immunology. 2020 ;11. Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2020.01624. [cited 2022 Aug 4].
Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X, et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci. 2021;17:3837–49.
Franke AJ, Skelton WP, Starr JS, Parekh H, Lee JJ, Overman MJ, et al. Immunotherapy for Colorectal Cancer: A Review of Current and Novel Therapeutic Approaches. J Natl Cancer Inst. 2019;111:1131–41.
Passardi A, Canale M, Valgiusti M, Ulivi P. Immune Checkpoints as a Target for Colorectal Cancer Treatment. International Journal of Molecular Sciences. Multidisciplinary Digital Publishing Institute; 2017;18:1324. Available from: https://www.mdpi.com/1422-0067/18/6/1324. [cited 2022 Apr 5].
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz H-J, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–91. Available from: https://www.sciencedirect.com/science/article/pii/S1470204517304229. [cited 2022 Apr 5].
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015;372:2509–20.
Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017;7:675–93. Available from: https://doi.org/10.1158/2159-8290.CD-17-0226. [cited 2022 Apr 6].
McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Overman MJ, Lonardi S, Wong KYM, Lenz H-J, Gelsomino F, Aglietta M, et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair–Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. JCO Wolters Kluwer. 2018;36:773–9. Available from: https://ascopubs.org/doi/10.1200/JCO.2017.76.9901. [cited 2022 Apr 6].
Lenz H-J, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-Line Nivolumab Plus Low-Dose Ipilimumab for Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: The Phase II CheckMate 142 Study. JCO Wolters Kluwer. 2022;40:161–70. Available from:https://ascopubs.org/doi/10.1200/JCO.21.01015. [cited 2022 Apr 6].
Le DT, Kim TW, Van Cutsem E, Geva R, Jäger D, Hara H, et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability–High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. JCO Wolters Kluwer. 2020;38:11–9. Available from: https://ascopubs.org/doi/10.1200/JCO.19.02107. [cited 2022 Apr 6].
André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N Engl J Med. Massachusetts Medical Society; 2020;383:2207–18. Available from: https://doi.org/10.1056/NEJMoa2017699. [cited 2022 Apr 6].
Gomez-Roca CA, Yanez E, Im S-A, Castanon Alvarez E, Senellart H, Doherty M, et al. LEAP-005: A phase 2 multicohort study of lenvatinib plus pembrolizumab in patients with previously treated selected solid tumors—Results from the colorectal cancer cohort. JCO Wolters Kluwer. 2021;39:3564–3564. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.3564. [cited 2022 Apr 15].
Ree AH, Hamre H, Kersten C, Hofsli E, Guren MG, Sorbye H, et al. Repeat sequential oxaliplatin-based chemotherapy (FLOX) and nivolumab versus FLOX alone as first-line treatment of microsatellite-stable (MSS) metastatic colorectal cancer (mCRC): Initial results from the randomized METIMMOX study. JCO Wolters Kluwer. 2021;39:3556–3556. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.3556. [cited 2022 Apr 15].
Fakih M, Raghav KPS, Chang DZ, Bendell JC, Larson T, Cohn AL, et al. Single-arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (pMMR)/microsatellite stable (MSS) colorectal cancer (CRC). JCO Wolters Kluwer. 2021;39:3560–3560. Available from:https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.3560. [cited 2022 Apr 15].
Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. 2021;21:769–84. Available from: https://www.nature.com/articles/s41577-021-00539-6. [cited 2022 May 16].
Wang Z, Cao YJ. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front Immunol. 2020;11:176.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Mullard A. FDA approves first CAR T therapy. Nat Rev Drug Discov. 2017;16:669.
Moreno V, Hernandez T, de Miguel M, Doger B, Calvo E. Adoptive cell therapy for solid tumors: Chimeric antigen receptor T cells and beyond. Current Opinion in Pharmacology. 2021;59:70–84. Available from: https://www.sciencedirect.com/science/article/pii/S1471489221000606. [cited 2022 May 16].
Zhang C, Wang Z, Yang Z, Wang M, Li S, Li Y, et al. Phase I Escalating-Dose Trial of CAR-T Therapy Targeting CEA+ Metastatic Colorectal Cancers. Mol Ther. 2017;25:1248–58. Available from: https://www.sciencedirect.com/science/article/pii/S1525001617301089. [cited 2022 Apr 17].
Magee MS, Abraham TS, Baybutt TR, Flickinger JC Jr, Ridge NA, Marszalowicz GP, et al. Human GUCY2C-Targeted Chimeric Antigen Receptor (CAR)-Expressing T Cells Eliminate Colorectal Cancer Metastases. Cancer Immunol Res. 2018;6:509–16. Available from: https://doi.org/10.1158/2326-6066.CIR-16-0362. [cited 2022 Apr 17].
Lonez C, Hendlisz A, Shaza L, Aftimos P, Vouche M, Donckier V, et al. Celyad’s novel CAR T-cell therapy for solid malignancies. Curr Res Transl Med. 2018;66:53–6. Available from: https://www.sciencedirect.com/science/article/pii/S2452318618300163. [cited 2022 Apr 17].
Shaza L, Hendlisz A, Awada A, Canon L, Carrasco J, Cutsem EV, et al. Results from the completed dose-escalation phase I SHRINK study evaluating the autologous NKG2D-based CAR T-cell therapy CYAD-01 in metastatic colorectal cancer patients. :1.
Prenen H, Rasschaert M, Hendlisz A, Shaza L, Alcantar E, Cerf E, et al. Results from the completed dose-escalation of the ALLOSHRINK phase I study evaluating the allogeneic NKG2D-based CAR T-cell therapy CYAD-101 in metastatic colorectal cancer patients. :1.
Kim RD, Prenen H, Rottey S, Kim DW, Flament A, Lehmann F, et al. KEYNOTE-B79 phase 1b trial to evaluate the allogeneic CAR T-cells CYAD-101 and pembrolizumab in refractory metastatic colorectal cancer patients. JCO. 2022;40:TPS227–TPS227. Available from: https://ascopubs.org/doi/10.1200/JCO.2022.40.4_suppl.TPS227. [cited 2022 Apr 18].
Jou J, Harrington KJ, Zocca M-B, Ehrnrooth E, Cohen EEW. The Changing Landscape of Therapeutic Cancer Vaccines—Novel Platforms and Neoantigen Identification. Clin Cancer Res. 2021;27:689–703. Available from: https://doi.org/10.1158/1078-0432.CCR-20-0245. [cited 2022 May 16].
Blass E, Ott PA. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat Rev Clin Oncol. 2021;18:215–29.
Sellars MC, Wu CJ, Fritsch EF. Cancer vaccines: Building a bridge over troubled waters. Cell. 2022;185:2770–88.
Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18:168–82.
Kamath V. Cancer vaccines: An unkept promise? Drug Discov Today. 2021;26:1347–52. Available from: https://www.sciencedirect.com/science/article/pii/S1359644621000726. [cited 2022 Aug 4].
Mougel A, Terme M, Tanchot C. Therapeutic Cancer Vaccine and Combinations With Antiangiogenic Therapies and Immune Checkpoint Blockade. Frontiers in Immunology. 2019;10. Available from: https://www.frontiersin.org/article/10.3389/fimmu.2019.00467. [cited 2022 May 16].
Ye T, Li F, Ma G, Wei W. Enhancing therapeutic performance of personalized cancer vaccine via delivery vectors. Adv Drug Deliv Rev. 2021;177:113927. Available from:https://www.sciencedirect.com/science/article/pii/S0169409X21003203. [cited 2022 Aug 4].
Li W-H, Li Y-M. Chemical Strategies to Boost Cancer Vaccines. Chem Rev. 2020;120:11420–78. Available from: https://doi.org/10.1021/acs.chemrev.9b00833. [cited 2022 Aug 4].
Vermorken JB, Claessen AM, van Tinteren H, Gall HE, Ezinga R, Meijer S, et al. Active specific immunotherapy for stage II and stage III human colon cancer: a randomised trial. The Lancet. 1999;353:345–50. Available from: https://www.sciencedirect.com/science/article/pii/S0140673698071864. [cited 2022 Apr 25].
Naqvi E. Immuno-oncology Therapy OncoVAX Is a Colon Cancer Vaccine. Available from: https://immuno-oncologynews.com/oncovax//. [cited 2022 Apr 25].
Sabado RL, Balan S, Bhardwaj N. Dendritic cell-based immunotherapy. Cell Res. 2017;27:74–95. Available from: https://www.nature.com/articles/cr2016157. [cited 2022 Aug 4].
Bol KF, Schreibelt G, Rabold K, Wculek SK, Schwarze JK, Dzionek A, et al. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J Immunother Cancer. 2019;7:109.
Wang Y, **ang Y, **n VW, Wang X-W, Peng X-C, Liu X-Q, et al. Dendritic cell biology and its role in tumor immunotherapy. J Hematol Oncol. 2020;13:107.
Wculek SK, Amores-Iniesta J, Conde-Garrosa R, Khouili SC, Melero I, Sancho D. Effective cancer immunotherapy by natural mouse conventional type-1 dendritic cells bearing dead tumor antigen. J Immunother Cancer. 2019;7:100.
Liu W, Tang H, Li L, Wang X, Yu Z, Li J. Peptide-based therapeutic cancer vaccine: Current trends in clinical application. Cell Prolif. 2021;54:e13025.
Stephens AJ, Burgess-Brown NA, Jiang S. Beyond Just Peptide Antigens: The Complex World of Peptide-Based Cancer Vaccines. Front Immunol. 2021;12:696791.
Snook AE, Baybutt TR, **ang B, Abraham TS, Flickinger JC, Hyslop T, et al. Split tolerance permits safe Ad5-GUCY2C-PADRE vaccine-induced T-cell responses in colon cancer patients. J Immunother Cancer. BMJ Specialist Journals; 2019;7:104. Available from: https://jitc.bmj.com/content/7/1/104. [cited 2022 Apr 23].
Hubbard JM, Cremolini C, Graham RP, Moretto R, Mitchelll JL, Wessling J, et al. A phase I study of PolyPEPI1018 vaccine plus maintenance therapy in patients with metastatic colorectal cancer with a predictive biomarker (OBERTO). JCO Wolters Kluwer. 2019;37:3557–3557. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.3557. [cited 2022 Apr 24].
Acknowledgements
The figures were created with BioRender.com. I would like to express my sincere gratitude to Prof. Dawei Li, from whose careful teaching I have benefited greatly in preparing for this review. I would also like to give special thanks to Prof. ** Wei, who has given me great help and encouragement.
Funding
This research was supported by grants from the National Natural Science Foundation of China (81972293, 81972185) and Dawn program of Shanghai Education Commission (21SG09).
Author information
Authors and Affiliations
Contributions
Yaxian Wang and **nyang Zhong had an equal contribution to this manuscript. Dawei Li and ** Wei designed and critically reviewed the manuscript. Yaxian Wang and **nyang Zhong drafted the manuscript. Xuefeng He, Zijuan Hu, Huixia Huang, Jiayu Chen and Keji Chen collected literature and completed tables. Senlin Zhao checked and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Y., Zhong, X., He, X. et al. Liver metastasis from colorectal cancer: pathogenetic development, immune landscape of the tumour microenvironment and therapeutic approaches. J Exp Clin Cancer Res 42, 177 (2023). https://doi.org/10.1186/s13046-023-02729-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-023-02729-7