
Abstract
Research into the potential benefits of artificial intelligence for comprehending the intricate biology of cancer has grown as a result of the widespread use of deep learning and machine learning in the healthcare sector and the availability of highly specialized cancer datasets. Here, we review new artificial intelligence approaches and how they are being used in oncology. We describe how artificial intelligence might be used in the detection, prognosis, and administration of cancer treatments and introduce the use of the latest large language models such as ChatGPT in oncology clinics. We highlight artificial intelligence applications for omics data types, and we offer perspectives on how the various data types might be combined to create decision-support tools. We also evaluate the present constraints and challenges to applying artificial intelligence in precision oncology. Finally, we discuss how current challenges may be surmounted to make artificial intelligence useful in clinical settings in the future.
Similar content being viewed by others


Introduction
In the upcoming decades, it is anticipated that cancer would surpass other illnesses as one of the main global causes of morbidity and mortality [1]. A recent study from The Lancet [2] demonstrated that for many low-income and middle-income nations, noncommunicable diseases (NCDs) pose an ever-greater health threat, with cancer becoming an NCD of greater importance. Therefore, it is imperative to focus on cancer treatment, enhance the rate of early detection and cure, and boost cancer screening.
Due to technical advancements in statistics and computer software, computer professionals, and health scientists may now collaborate closely to improve prognoses. As a result of the adoption of artificial intelligence (AI) strategies, researchers have increasingly concentrated on creating models using AI algorithms to detect and diagnose cancer. AI is the process of teaching a computer to mimic human intelligence by showing it how to study, evaluate, comprehend, deduce, interact, and make decisions [3]. Tremendous success has been achieved with AI in the last ten years in the fields of speech synthesis, natural language processing, and computer vision. This review focuses on the latest AI techniques for tumor diagnosis, treatment, and prognosis. We highlight artificial intelligence applications for omics data types, and we offer perspectives on how the various data types might be combined to create decision-support tools and discuss how current challenges may be surmounted to make artificial intelligence useful in clinical settings in the future.
We searched three databases from their creation until November 10, 2023: MEDLINE (PubMed), CENTRAL (Cochrane Central Register of Controlled Trials), and Embase to assess the published literature pertaining to the application of artificial intelligence in cancer. Due to the rapid pace of AI updates, we have focused on the last two years of relevant research. The following keywords were used in this sco** review: (neoplasms OR cancer) AND (artificial intelligence OR deep learning OR machine learning). With a focus on the application and usage of artificial intelligence in cancer treatment, we incorporated a total of 254 publications in the construction of this narrative review, including pertinent prospective, retrospective, and review studies.
Specific meaning of artificial intelligence
AI is an area of computer technology comprising numerous techniques and subfields aimed at performing activities that could previously be completed only by humans. To enhance the interpretation of medical data relevant to medical administration, diagnostics, and predictive outcomes, AI technologies and their subdomains are being implemented in healthcare delivery. The two main techniques for implementing AI are machine learning (ML) and deep learning (DL), which are terms that are frequently used interchangeably. Deep learning is a branch of machine learning. ML generates predictions by spotting patterns in data by means of mathematical algorithms. DL produces forecasts using multiple layers of fabric neural network algorithms that are modeled after the brain’s neural network architecture. In the past ten years, with advancements in big data, algorithms, computing power, and Internet technology, AI has excelled in numerous tasks across a wide range of industries, including identification of faces, image classification, speech recognition, automatic translation, and healthcare [4]. The main ML techniques are support vector machines (SVMs), decision trees, and K unsupervised algorithms, while the most commonly used for DL today are convolutional neural networks (CNNs) [5]. Figure 1 presents a few of the most basic ML and DL approaches.

Network structure of DL. a A model of an SVM; b A model of a random forest that is composed of several decision trees; c KNN characterized by the fact that it is composed of many random features rather than a linear feature; d Components of CNNs [6]; and e Components of graphical CNNs
ML fundamentally seeks to replicate or mimic humans’ capacity for pattern recognition. Traditional ML approaches take far longer to teach and test based on a specific problem than DL approaches. SVMs, decision trees, random forests, gradient boosting (such as XGBoost), and other conventional ML techniques are examples of traditional ML techniques. There is a significant flaw with decision trees, namely a decision tree divides samples extremely precisely, but dividing samples too precisely causes overfitting of the training set, and dividing samples coarsely results in a decision tree that does not fit the samples properly. Decision trees called random forests are based on the concept of learning and bagging combined. Two factors—the random selection of the dataset and the random selection of the characteristics utilized in each tree—reflect the unpredictability of a random forest the most. The XGBoost technique repeatedly constructs an ensemble of decision trees. The capacity of this technique to manage missing data, capture nonlinear correlations between the model features and the outcome, and have higher-order interactions between variables is its key benefits over conventional logistic regression-based risk models [7].
Training artificial intelligence models
Several processes are necessary for training an AI model, including data gathering and preparation, model selection, model training, and hyperparameter tuning.
Data collection and preprocessing
With the rapid development of modern medicine, various types of data are emerging. A large amount of imaging data has been generated as represented by X-ray, CT, and MRI, and the development of pathology has made sectioning the gold standard for tumor diagnosis. In addition to the traditional clinical information data, with the remarkable advances in sequencing technology over the past two decades, how to deal with the large amount of molecular data brought about by genomics, transcriptomics, proteomics, etc., has also become a matter of close attention for clinicians. Later, we will describe how to deal with a single type of data. However, a patient usually does not have only one type of test and one type of data, so we will also introduce how to integrate different types of data to enhance computational models.
To facilitate the subsequent model training, we need to preprocess these data. For digital data, we need to remove outliers, deal with missing values, and normalize the data. The most often utilized AI algorithms, using EHR as an example, are deep learning, decision tree algorithms, and regression algorithms. While completing regression tasks to finish disease risk prediction, researchers also use classification tasks to extract lesion characteristics from illnesses and categorize them [45, 46]. A DL system [47] was created that can correctly identify the existence of lung cancer within three years and account for all pertinent nodule and nonnodule markers on screening chest CTs. Their research was the first to create a deep machine learning prediction method without the use of computer-aided diagnostic tools to assess a person’s 3-year probability of develo** lung cancer and related lung cancer-specific mortality. Kiran Vaidhya Venkadesh et al. [48] created and externally verified a CNN-based DL algorithm for estimating the likelihood of malignancy in lung nodules found by low-dose screening CT, which demonstrated good performance, on par with thoracic radiologists, at estimating the malignancy risk of pulmonary nodules observed during screening CT(AUC = 0.93). However, their researches included a number of restrictions. Firstly, one CT scan was employed in the created method, and a prior CT image was not taken into account [48]. Secondly, on average, members of the cohort [47] had LDCTs for screening every year, which may cause bias in the measurement results.
In order to address the above issues, a deep learning system was developed that can forecast the probability of develo** lung cancer six years from now. Newly developed Sybil [90] to classify skin lesions and to present dermatologist-level prediction outcomes. The knowledge distillation approach is also often used to help diagnose melanoma [91, 92]. In addition to the simple teacher–student model, the SSD-KD approach [93], a unique self-supervised diversified knowledge distillation technique, has been used for the lightweight multiclass categorization of skin diseases utilizing dermoscopy images. In that study, the conventional single relational modeling block was substituted with dual relational blocks in terms of technological innovation. Multi-Site Cross-Organ Calibrated Deep Learning (MuSClD), a novel approach to cross-organ calibration between two sites of digitalized histopathology images, was validated in nonmelanoma skin cancer. 3D images [94, 95], EfficientNet [96, 97], genetic programming (GP) [98], and new AI algorithms on smartphones [99, 100] have also been developed for skin cancer diagnosis.
To supplement human visual inspection, AI can assist in the detection of undetectable tumor lesions on PET scans. Ga-PSMA-11 PET-based radiomics features have been used to generate random forest models that accurately predicted invisible intraprostatic lesions [101]. Biopsy and magnetic resonance imaging (MRI) are frequently used to diagnose intracranial tumors. Due to the similar phenotypes of various tumor classes on MRI scans, it has been difficult to identify tumor types, especially rare types, from MRI data. A DL method for segmenting and classifying 18 distinct types of intracranial tumors was developed [102] using T1- and T2-weighted images and T2 contrast MRI sequences and evaluated with an AUC of 0.92.
AI may easily be applied to medical imaging, and major advancements in this area have been made in recent years. AI eliminates the uncertainty that people contribute to decisions and delivers objective measurements for each choice. However, the limits are also readily apparent. The molecular causes of illnesses are not revealed by morphological evidence. By using this method, disease states with the same morphological appearance cannot be discriminated.
Tumor staging and grading
Important factors for tumor T-staging include the size and degree of invasiveness of primary tumors, which comprise descriptions of their shapes. Convolutional neural networks are most used in this task. The T stage of Barrett’s carcinoma is a crucial consideration when choosing a course of therapy. Endoscopic ultrasonography is still the norm for preoperative staging, but its usefulness is under question. To help with staging and to improve outcomes, new tools are needed. With a high accuracy of 73% in diagnosing esophageal cancer, an AI system built around endoscopic images has been developed [103]. Tumor sizes and forms vary, making individual slice-by-slice screening for T-staging time intensive. Consequently, a multi-perspective aggregation network (TSD Net) has been created with ideas from oncological diagnostics that included different diagnosis-oriented knowledge and enabled automatic nasopharyngeal carcinoma T-staging identification [104].
Advances in imaging histology have greatly contributed to hel** TNM staging of tumors. Separate iterations of the machine learning models have been created using both the entire collection of extracted features (full model) and just a selection of the previously discovered robust metrics (robust models) to confirm that CT-based radiomics signatures were effective tools for determining the grade and stage of ccRCC [105]. Additionally important in the early phases of decision-making, but time-consuming, is a delineation of the tumor. To forecast the grade of a tumor while also segmenting it, a single multi-task convolutional neural network has been created using the whole 3D, structural, preoperative MRI data [106].
Accurate assessment of lymph node metastasis (LNM) is essential for evaluating the staging and grading of tumor patients. In addition to offering a straightforward “yes” or “no” response on the likelihood of having cancer, AI models can also identify the disease site from a test picture. One of the most common applications is to help find the localization of metastatic tumors. Using whole-body PET/CT scans, convolutional neural networks (CNNs) based on UNet [1] were trained to detect and separate metastatic prostate cancer lesions fully automatically [107]. The localization of tumor metastasis in whole-slide images has also been studied extensively in recent years [107,108,109,110]. The condition of the lymph nodes (LNs) prior to surgery is crucial for the management of colorectal cancer (CRC). With areas under the curve (AUCs) of 0.79, 0.73, and 0.70 in the training set, testing set, and verification set, respectively, a deep learning (DL) model [111] with features gathered from improved venous-phase CT images of CRC has been proposed to identify LNM in CRC. Shaoxu Wu et al. [112] created a diagnostic algorithm called LNMDM based on AI that was effective for finding micrometastases in lymph nodes and was demonstrated not only in bladder cancer (0·983 [95% CI 0·941–0·998]) but also in breast cancer (0·943 [95% CI 0·918–0·969]) and prostate cancer (0·922 [95% CI 0·884–0·960]). AI plays a significant role in aiding diagnostics to find lymph node metastases in slide pictures. Lymph node metastases, especially micrometastases, were successfully identified by the LNMDM [112] on whole-slide images in bladder cancer. The VIS AI algorithm demonstrated comparable accuracy and NPV in identifying LN metastases on breast cancer. In summary, the implementation of AI in tumor staging and grading has significantly improved tumor prognoses and increased the general survival rate of cancer patients.
Tumor therapy
AI for exploring tumor therapeutic targets
In recent years, the development of multiomics technologies in cancer research [113, 114] has greatly facilitated the discovery of anticancer targets [115,116,117]. The advancement of precision medicine and translational medicine will be significantly aided by the use of ML and DL to mine multiomics data to investigate complicated disease causation processes and treatment response mechanisms. In the following, we describe in detail the advances in genomics, epigenetics, transcriptomics, proteomics, metabolomics, and multiomics in cancer target discovery. Figure 3 describes the main sources of these six components and the advanced methods currently comprising them.

Components of multiomics and the main techniques. The combination of AI and multiomics has led to the discovery of new targets for cancer therapy. (Created with BioRender.com)
Genomics
The genome contains inherited information that controls gene expression to shape the structure and working machinery of the cell [118]. Genomics focuses on understanding the composition, organization, visualization, and modification of an organism’s whole genome [119]. The rise of the genomic era has also boosted precision medicine and cancer [120]. The approach of a meta-learning model [121] allows users to discover significant pathways in cancer and priority genes based on their contribution to survival prediction. To fully understand how cancer develops, progresses, and is treated, accurate somatic mutation detection is difficult yet essential. The first method for detecting somatic mutations based on deep CNNs is called NeuSomatic [122]. However, the fact that matched normal specimens are not frequently acquired in clinical practice is a major barrier to genetic testing in cancer. The somatic vs. germline status of each discovered change may be predicted using SGZ, [123] which does not need a patient-related standard control, by modeling the mutation’s allele frequency (AF), accounting for the cancer content, cancer ploidy, and local copy number. Similarly, a recently created method, Continuous Representation of Codon Switches [124] (CRCS), a DL-based technique, can aid in the identification and investigation of driver genes as well as the detection of cancer-related somatic mutations in the absence of matched normal samples.
Taking colon cancer as an example, numerous studies [125,126,127,128] have subtyped colorectal cancer based on similar and different biological traits and pathways, and they have identified the relationships between these pathways and patient prognosis, overall survival, and responsiveness to various treatments—particularly targeted therapy and immunotherapy. Using 499 primary colorectal neoplasm diagnostic images from 502 individuals in The Cancer Genome Atlas Colon and Rectal Cancer (TCGA-CRC-DX) cohort, a retrospective study established a weakly supervised DL framework incorporating three separate CNN models [85]. After comprehensive validation, the method was shown to be helpful for patient classification for targeted medicines, with possible cost savings and quicker turnaround times compared to sequencing- or immunohistochemistry-based techniques. The research, however, examined each individual image tile without considering the significance of the spatial relationship between tiles. In a recent study [129], a method for forecasting cross-level molecular profiles involving gene mutations, copy number variations, and functional protein expression from whole-slide pictures was proposed. This method focuses on the spatialization of cancer tiles. In the training dataset, the model performed exceptionally well in predicting a variety of genetic alterations and then identifying targeted therapies for colon cancer patients.
Epigenetics
Epigenetic modification is the genetic change in the way genes operate and express without altering the DNA sequence. DNA methylation, histone modification, and chromatin structure manipulation are the three primary epigenetic modifications that are now understood [130]. Although there are high-quality data on DNA methylation, few samples have RNA-seq data due to numerous experimental difficulties. Therefore, an innovative technique called TDimpute [131] was created to reconstruct lost data on gene expression from DNA methylation data using a transfer learning-based neural network. Understanding how epigenetics regulates gene expression to govern cell functional heterogeneity is dependent on the ability to predict differentially expressed genes (DEGs) from epigenetic signal information. On the basis of epigenetic data, a multiple self-attention model (Epi-MSA) [132] was suggested to predict DEGs. To determine which gene locations are crucial for forecasting DEGs, Epi-MSA first applies CNNs for neighborhood bin information embedding and then makes use of several self-attention encoders on various input epigenetic parameters.
Transcriptomics
Transcriptomics is a useful tool for comprehending the physiology of cancer and locating biomarkers. It includes analyses of alternative transcription and alternative polyadenylation, detection of integration transcripts, investigations of noncoding RNAs, transcript annotation, and finding novel transcripts [133]. One study using DL algorithms to interpret common cancer transcriptome markers [134] showed that across a wide range of solid tumor types, dysregulation of RNA-processing genes and aberrant splicing are widespread traits on which fundamental cancer pathways may converge. Molecular pathology plays an important role in cancer, but whether it is possible to estimate the levels of gene expression based on a visual inspection of H&E-stained WSIs has never been thoroughly explored. Numerous studies have been conducted to predict cancer gene expression, including that of prostate [135] and breast [136] cancers, across the transcriptome from histopathological images. A DL model called HE2RNA [137] based on a multitasking poorly supervised technique was created using matched WSIs and RNA-Seq profiles from TCGA data, which included 8725 patients and 28 distinct cancer types. This increases the likelihood of discovering novel gene targets. Patients’ responses to treatment are significantly influenced by the quantity, composition, and geographic distribution of the cell groups in the tumor microenvironment (TME) [138]. The thorough characterization of gene regulation in the TME has been made possible by recent developments in spatial transcriptomics (ST) [139, 140]. Three new approaches have recently been developed: Kassandra [141], XFuse [142], and TESLA [143]. Kassandra is a tree ML algorithm that was taught to precisely rebuild the tumor microenvironment (TME) using a large database of > 9,400 tissue- and blood-sorted cell RNA profiles combined into millions of artificial transcriptomes. According to Kassandra’s deconvolution of TME components, these populations play a part in tumor etiology and other biological processes. By utilizing data from H&E-stained histological images, XFuse predicts superresolution gene expression per pixel. TESLA is an ML framework that incorporates gene expression and histological image data into ST to study the TME. The innovative aspect of TESLA is the annotation of diverse immune and tumor cells on histological images directly.
In addition, the identification of lncRNAs [144,145,146] and microRNAs [147, 148] by ML can assist in the precise treatment of cancer. In the fight against cancer, therapeutic decisions are increasingly based on molecular tumor features, and cancer tissue molecular profiling is becoming an essential component of standard diagnosis [149]. To reduce individualized patient differences, scGeneRAI [150] uses layerwise relevance propagation (LRP), an explainable AI technique, to extrapolate individual cell gene regulation networks from single-cell RNA sequencing data. Oncology drug response is a major challenge in cancer treatment. With an average Matthew correlation coefficient (MCC) and AUC of 0.56 and 0.80, respectively, the classification and regression tree (CART) model from interpretable ML models has proven to be the best model for predicting how breast cancer would react to doxorubicin [151]. At the single-cell level, ScDEAL is a deep transfer learning system that integrates bulk cell-line data to predict cancer medication response at the single-cell level. Finding drug resistance targets at the level of transcriptional profiles using AI deserves more research in the future.
Proteomics
Proteomics is a broad study of proteins that identifies and counts the proteins present in a biological sample, such as a sample of cells, tissues, or bodily fluids. Proteomics data offer the benefit of providing a numerical number of individual proteins throughout the body and dynamic characteristics that develop over time and among individual subjects, in contrast to other forms of omics data, such as genomic data. Mass spectrometry (MS) is a key tool used in proteomics research [125]. MS-based proteomics has advanced quickly in terms of lower cost and higher throughput, regularly permitting large-cohort studies with tens of thousands of participants and tens of millions of identified proteins in cancer cells and other biological samples. However, the majority of research concentrates on the final proteins discovered using a collection of algorithms that compare partial MS spectra with the ordered database, leaving the problem of pattern identification and categorization of the raw mass-spectrometric information unanswered. Consequently, for the analysis of massive MS data using deep neural networks (DNNs), the publicly available MSpectraAI [152] platform and the tumor classifier [153] have been developed, which could expand the intriguing use of DL techniques for classifying and predicting proteomics data from multiple cancer types and distinguishing between tumor and nontumor samples.
Sequential Window Acquisition of all Theoretical Mass Spectra-MS (SWATH-MS) is a cutting-edge MS method that enables the measurement of nearly all peptides as well as proteins present in a single sample, making it valuable in research involving massive sample cohorts [154]. It can be used to facilitate the categorization of CRC molecular subgroups and promote both diagnostics and the creation of novel medications [155]. Regarding colorectal cancer, a mechanism-based ML approach [156] has been proposed to find genes and proteins with substantial correlations to event-free patient survival and predictive potential to account for patient-specific variations in STN activity by building three linear regression models. The development of proteomics has contributed to the discovery of new targets in hematological tumors. Targetable enzyme characteristics have been revealed by proteomics of acute lymphoblastic leukemia that is resistant to Notch1 suppression [157]. Through the induction of long-lasting immune responses, T cells play critical roles in human defense against hematological tumors. In recent work [158], ML and nanoscale proteomics were coupled to subtype T cells in peripheral bloodstreams from single individuals with multiple myeloma. To reduce the possibility of overfitting the ML models, differentially expressed proteins (DEPs) were selected according to statistical significance, and only the top 13–15 DEPs were utilized. Thus, this work helped identify new targets for immunotherapy. Another DL network [159] identified the 20 proteins most strongly associated with FLT3-ITD in acute myeloid leukemia. In addition, DL and ML have been applied to proteomics data for pancreatic cancer [160] and diffuse large B-cell lymphoma [161] patients, respectively.
Metabolomics
Metabolomics is a burgeoning area of research that utilizes technologically sophisticated analytical chemistry to perform high-throughput characterization of metabolites in cells, organs, tissues, or biological fluids [162]. New therapeutic targets have been suggested to target metabolic constraints in cancer as a result of metabolomics studies, which have revealed potential medicinal weak points for treating cancer [163]. Lipidomics is a branch of metabolomics that aims to study and analyze the lipids in the metabolome and the molecules that interact with them [164]. Metabolomics analysis can be performed using GC‒MS and LC‒MS, and LC‒MS is commonly used for the analysis of lipidomics. The combination of metabolomics and AI has flourished in various areas of cancer, including breast cancer [165, 166], head and neck cancer [167], colorectal cancer [168, 169], glioma cancer [170], esophageal cancer [171, 172], lung cancer [249]. The most susceptible source of information determines the total security level when we combine patient data from other sources. Clinical data are frequently the property of particular institutions due to concerns about patient privacy, and there are few methods in place to share data among institutions. It is frequently inadequate to remove personal identifiers and secret information since an attacker can still draw conclusions to retrieve some of the missing data. The good news is that multicenter information transfer agreements and safeguarding privacy distributed DL (DDL) are starting to overcome this roadblock [250,251,252]. DDL offers a mechanism that protects privacy so that several users can collaborate on learning using a deep model without directly exchanging local datasets. In addition, it is important to ascertain the level of supervision that doctors must provide and identify the person accountable for any poor choices made by DL tools. On the other hand, we should educate AI users to guarantee that they are knowledgeable consumers of the technology and endeavor to openly and clearly express to them what they should anticipate in a variety of circumstances. Many of the hazards described above may be reduced by being accessible, having varied demands, and being cautious.
When implementing AI, ethical issues are crucial since unethical data gathering or usage practices might introduce biases into models. These biases can take numerous forms, but they are mostly determined by the data and cohort composition employed by the particular AI systems. Providing and reviewing AI models lacks defined criteria or norms. Identifying the possible biases included in the established systems will be crucial; thus, future studies should fill this knowledge gap to help researchers and physicians.
Clinical integration
As mentioned above, AI has been shown in many studies to improve the correctness of cancer diagnosis. However, a different perspective has been proposed. In one study [253], the authors systematically evaluated 131 published studies using the QUality Assessment of Diagnostic Accuracy Studies-2 (QUADAS-2) tool. They reported that the accuracy of AI in breast cancer screening programs cannot currently be evaluated based on available research, and it is unclear where in the therapeutic pathway AI could be most helpful.
For LLMs, one of their advantages is the capacity to sift through vast volumes of data and provide replies in a conversational and understandable manner. LLMs also have the potential to be used in patient education and consultation, offering patient-friendly information to aid in their understanding of their medical issues and available treatment choices, facilitating joint decision-making. More crucially, LLMs can contribute to the democratization of medical knowledge by allowing anybody, regardless of location or socioeconomic position, quick access to reliable medical information. However, special attention needs to be paid to the fact that current LLMs are not yet capable of fully replacing doctors, as they may contain errors or omit key points in the responses. Although ChatGPT-4.0 was more accurate than the other tools, neither ChatGPT nor Google Bard or the Bing or Google search engines provided 100% accurate answers to all queries [242]. The much-anticipated Med-PaLM, while promising, is evaluated by multiple choice questions; however, real life is not multiple choice, and different clinical symptoms and specificities in different patients make clinical diagnosis more complex. While AI, such as ChatGPT-4.0, might be helpful for giving broad information and responding to frequently asked queries. Nonetheless, it is important to take great caution when responding to inquiries from certain patients. It is essential to continuously upgrade AI models to include the most recent medical information.
Currently, almost all relevant AI models have been created to assist in cancer diagnosis using clinical data from the time of development. These clinical data may be derived from patient reports, complaints, or sequencing results. The question is whether there is an AI model that can recommend more tests and treatment modalities or perhaps aid in prescribing anticancer medication without relying on clinical data. The current state of affairs is that with the development of multiomics, a variety of data, such as methylation and fragmentomics [254], are being used to train AI models. If one day the data of the AI model accumulates to a large enough size, is it possible to predict the probability of cancer occurrence by only entering the data of normal people, and is it possible to give the corresponding chemotherapy regimen by only comparing the sequencing results of cancer patients and the database. This is a question worth thinking about and very interesting. First, the database must be large enough and ethical; second, there is variability between individuals, and it would be irresponsible to treat them by looking only at sequencing data at the genetic level or transcriptional level, for example.
However, if it is only in the area of cancer diagnosis, AI models have the potential to identify molecules and biomarkers associated with mutated genes and thus confirm the diagnosis of cancer independently of traditional pathology measurements. Meanwhile, with the advent of wearable and portable medical instruments, AI has shown much potential for the early screening of tumors. Therefore, we think that in the future, AI models have the potential to impact the cancer diagnostic market, but in terms of treatment, they cannot be separated from doctors and clinical data.
What must be realized is that despite the rapid development and promising future of AI, it can never replace clinicians and will only become an important tool to assist them in the future.
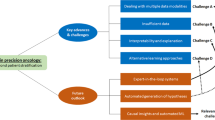
Conclusion
In summary, AI has the ability to fundamentally alter cancer treatment and move it closer to the promise of precision oncology. In an era where genomics is being incorporated into health delivery and health data are being more digitized, it is anticipated that AI would be used in the construction, verification, and application of decision-support tools to promote precision oncology. We highlighted several promising AI applications in this review, including detection, prognosis, and administration of cancer treatments. It is undeniable that large language model can greatly assist physicians in their clinical work, but it can never replace them. Important conditions for the general adoption of AI in clinical settings include phenotypically rich data for the development of models and clinical validation of the biological value of AI-generated insights. Finally, clinical validation of AI is required before it may be used in ordinary patient treatment.
Availability of data and materials
Not applicable.
Abbreviations
- AI:
-
Artificial Intelligence
- AUC:
-
Area under the curve
- CC:
-
Colon cancer
- ccRCC:
-
Clear cell renal cell carcinoma
- CDSS:
-
Clinical decision-support systems
- cGANs:
-
Conditional generative adversarial networks
- CNNs:
-
Convolutional neural networks
- CRC:
-
Colorectal cancer
- CT:
-
Computed tomography
- DBT:
-
Digital breast tomosynthesis
- DDL:
-
Distributed deep learning
- DEGs:
-
Differentially expressed genes
- DEPs:
-
Differentially expressed proteins
- DFS:
-
Disease free survival
- DGCN:
-
Directed Graph convolutional network
- DL:
-
Deep learning
- DNNs:
-
Deep neural networks
- EHR:
-
Electronic health record
- GBM:
-
Glioblastoma multiforme
- GCN:
-
Graph convolutional network
- GPT:
-
Generative pretrained transformers
- H&E:
-
Hematoxylin and eosin
- HNC:
-
Head and neck cancer
- KIRC:
-
Kidney renal clear cell carcinoma
- KNN:
-
K-Nearest neighbor
- LDCT:
-
Low-dose computed tomography
- LLMS:
-
Large language models
- LNM:
-
Lymph node metastasis
- ML:
-
Machine learning
- MLSL:
-
Multilabel softmax loss
- MRI:
-
Magnetic resonance imaging
- MS:
-
Mass spectrometry
- MSI:
-
Microsatellite instability
- NCDs:
-
Noncommunicable diseases
- NLP:
-
Natural language processing
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PET:
-
Positron emission tomography
- RSF:
-
Random survival forest
- ST:
-
Spatial transcriptomics
- SVM:
-
Support vector machine
- TCGA:
-
The Cancer Genome Atlas
- TME:
-
Tumor microenvironment
- TNM:
-
Tumor node metastasis
- USMLE:
-
United States Medical Licensing Examination
- WFO:
-
Watson for Oncology
- WSI:
-
Whole-slide image
References
Bray F, Jemal A, Grey N, Ferlay J, Forman D. Global cancer transitions according to the human development index (2008–2030): a population-based study. Lancet Oncol. 2012;13(8):790–801.
The L. Global cancer: overcoming the narrative of despondency. Lancet (London, England). 2023;401(10374):319.
Moor J. The Dartmouth College artificial intelligence conference: the next fifty years. AI Mag. 2006;27(4):87–91.
Yu KH, Beam AL, Kohane IS. Artificial intelligence in healthcare. Nat Biomed Eng. 2018;2(10):719–31.
Deo RC. Machine learning in medicine. Circulation. 2015;132(20):1920–30.
Zhou P, Cao Y, Li M, Ma Y, Chen C, Gan X, et al. HCCANet: histopathological image grading of colorectal cancer using CNN based on multichannel fusion attention mechanism. Sci Rep. 2022;12(1):15103.
Gould MK, Huang BZ, Tammemagi MC, Kinar Y, Shiff R. Machine learning for early lung cancer identification using routine clinical and laboratory data. Am J Respir Crit Care Med. 2021;204(4):445–53.
Liang J, He Y, **e J, Fan X, Liu Y, Wen Q, et al. Mining electronic health records using artificial intelligence: Bibliometric and content analyses for current research status and product conversion. J Biomed Inform. 2023;146: 104480.
Zhang G, Jiang Z, Zhu J, Dai T, He X, Liu X, et al. Innovative integration of augmented reality and optical surface imaging: a coarse-to-precise system for radiotherapy positioning. Med Phys. 2023;50(7):4505–20.
Yao Y, He L, Mei L, Weng Y, Huang J, Wei S, et al. Cell damage evaluation by intelligent imaging flow cytometry. Cytometry Part A : J Int Soci Anal Cytol. 2023;103(8):646–54.
DiSpirito A 3rd, Vu T, Pramanik M, Yao J. Sounding out the hidden data: a concise review of deep learning in photoacoustic imaging. Exp Biol Med (Maywood). 2021;246(12):1355–67.
Silver FH, Mesica A, Gonzalez-Mercedes M, Deshmukh T. Identification of cancerous kin lesions using vibrational optical coherence tomography (VOCT): use of VOCT in conjunction with machine learning to diagnose skin cancer remotely using telemedicine. Cancers. 2022;15(1):156.
Pérez-Cota F, Martínez-Arellano G, La Cavera III S, Hardiman W, Thornton L, Fuentes-Domínguez R, et al. Classification of cancer cells at the sub-cellular level by phonon microscopy using deep learning. Sci Rep. 2023;13(1):16228.
Niehues JM, Quirke P, West NP, Grabsch HI, van Treeck M, Schirris Y, et al. Generalizable biomarker prediction from cancer pathology slides with self-supervised deep learning: a retrospective multi-centric study. Cell reports Medicine. 2023;4(4): 100980.
Rönnau MM, Lepper TW, Amaral LN, Rados PV, Oliveira MM. A CNN-based approach for joint segmentation and quantification of nuclei and NORs in AgNOR-stained images. Comput Methods Programs Biomed. 2023;242: 107788.
Balasubramaniam S, Velmurugan Y, Jaganathan D, Dhanasekaran S. A modified LeNet CNN for breast cancer diagnosis in ultrasound images. Diagnostics (Basel, Switzerland). 2023;13(17):2746.
Tang Z, Li Z, Hou T, Zhang T, Yang B, Su J, et al. SiGra: single-cell spatial elucidation through an image-augmented graph transformer. Nat Commun. 2023;14(1):5618.
Azad R, Kazerouni A, Heidari M, Aghdam EK, Molaei A, Jia Y, et al. Advances in medical image analysis with vision transformers: a comprehensive review. Med Image Anal. 2023;91: 103000.
Li X, Fang X, Yang G, Su S, Zhu L, Yu Z. TransU2-Net: an effective medical image segmentation framework based on transformer and U2-Net. IEEE J Transl Eng Health Med. 2023;11:441–50.
Dascalu A, David EO. Skin cancer detection by deep learning and sound analysis algorithms: a prospective clinical study of an elementary dermoscope. EBioMedicine. 2019;43:107–13.
Walker BN, Rehg JM, Kalra A, Winters RM, Drews P, Dascalu J, et al. Dermoscopy diagnosis of cancerous lesions utilizing dual deep learning algorithms via visual and audio (sonification) outputs: laboratory and prospective observational studies. EBioMedicine. 2019;40:176–83.
Baltrusaitis T, Ahuja C, Morency LP. Multimodal machine learning: a survey and taxonomy. IEEE Trans Pattern Anal Mach Intell. 2019;41(2):423–43.
Mei X, Lee HC, Diao KY, Huang M, Lin B, Liu C, et al. Artificial intelligence-enabled rapid diagnosis of patients with COVID-19. Nat Med. 2020;26(8):1224–8.
Akselrod-Ballin A, Chorev M, Shoshan Y, Spiro A, Hazan A, Melamed R, et al. Predicting breast cancer by applying deep learning to linked health records and mammograms. Radiology. 2019;292(2):331–42.
Zhang K, Liu X, Xu J, Yuan J, Cai W, Chen T, et al. Deep-learning models for the detection and incidence prediction of chronic kidney disease and type 2 diabetes from retinal fundus images. Nature Biomed Eng. 2021;5(6):533–45.
Zhou HY, Yu Y, Wang C, Zhang S, Gao Y, Pan J, et al. A transformer-based representation-learning model with unified processing of multimodal input for clinical diagnostics. Nat Biomed Eng. 2023;7(6):743–55.
Moor M, Banerjee O, Abad ZSH, Krumholz HM, Leskovec J, Topol EJ, et al. Foundation models for generalist medical artificial intelligence. Nature. 2023;616(7956):259–65.
Faiella E, Vertulli D, Esperto F, Cordelli E, Soda P, Muraca RM, et al. Quantib prostate compared to an expert radiologist for the diagnosis of prostate cancer on mpMRI: a single-center preliminary study. Tomography (Ann Arbor, Mich). 2022;8(4):2010–9.
Eloy C, Marques A, Pinto J, Pinheiro J, Campelos S, Curado M, et al. Artificial intelligence-assisted cancer diagnosis improves the efficiency of pathologists in prostatic biopsies. Virchows Archiv : Int J Pathol. 2023;482(3):595–604.
Wang JY, Qu V, Hui C, Sandhu N, Mendoza MG, Panjwani N, et al. Stratified assessment of an FDA-cleared deep learning algorithm for automated detection and contouring of metastatic brain tumors in stereotactic radiosurgery. Radiat Oncol (London, England). 2023;18(1):61.
Seager A, Sharp L, Hampton JS, Neilson LJ, Lee TJW, Brand A, et al. Trial protocol for COLO-DETECT: a randomized controlled trial of lesion detection comparing colonoscopy assisted by the GI Genius™ artificial intelligence endoscopy module with standard colonoscopy. Colorectal disease: The Off J Assoc Coloproctol Great Britain and Ireland. 2022;24(10):1227–37.
Glissen Brown JR, Mansour NM, Wang P, Chuchuca MA, Minchenberg SB, Chandnani M, et al. Deep learning computer-aided polyp detection reduces adenoma miss rate: a United States Multi-center randomized tandem colonoscopy study (CADeT-CS Trial). Clin Gastroenterol Hepatol: Off Clin Pract J Am Gastroenterol Assoc. 2022;20(7):1499-507.e4.
Eden KB, Ivlev I, Bensching KL, Franta G, Hersh AR, Case J, et al. Use of an online breast cancer risk assessment and patient decision aid in primary care practices. J Women’s Health. 2020;29(6):763–9.
Niehoff JH, Kalaitzidis J, Kroeger JR, Schoenbeck D, Borggrefe J, Michael AE. Evaluation of the clinical performance of an AI-based application for the automated analysis of chest X-rays. Sci Rep. 2023;13(1):3680.
Kung TH, Cheatham M, Medenilla A, Sillos C, De Leon L, Elepaño C, et al. Performance of ChatGPT on USMLE: potential for AI-assisted medical education using large language models. PLOS digital health. 2023;2(2): e0000198.
Conant EF, Barlow WE, Herschorn SD, Weaver DL, Beaber EF, Tosteson ANA, et al. Association of digital breast tomosynthesis vs digital mammography with cancer detection and recall rates by age and breast density. JAMA Oncol. 2019;5(5):635–42.
Hofvind S, Holen ÅS, Aase HS, Houssami N, Sebuødegård S, Moger TA, et al. Two-view digital breast tomosynthesis versus digital mammography in a population-based breast cancer screening programme (To-Be): a randomised, controlled trial. Lancet Oncol. 2019;20(6):795–805.
Pattacini P, Nitrosi A, Giorgi Rossi P, Iotti V, Ginocchi V, Ravaioli S, et al. Digital mammography versus digital mammography plus tomosynthesis for breast cancer screening: the reggio emilia tomosynthesis randomized trial. Radiology. 2018;288(2):375–85.
Dang PA, Freer PE, Humphrey KL, Halpern EF, Rafferty EA. Addition of tomosynthesis to conventional digital mammography: effect on image interpretation time of screening examinations. Radiology. 2014;270(1):49–56.
Shoshan Y, Bakalo R, Gilboa-Solomon F, Ratner V, Barkan E, Ozery-Flato M, et al. Artificial intelligence for reducing workload in breast cancer screening with digital breast tomosynthesis. Radiology. 2022;303(1):69–77.
Nam JG, Hwang EJ, Kim J, Park N, Lee EH, Kim HJ, et al. AI Improves nodule detection on chest radiographs in a health screening population: a randomized controlled trial. Radiology. 2023;307(2): e221894.
Sim Y, Chung MJ, Kotter E, Yune S, Kim M, Do S, et al. Deep convolutional neural network-based software improves radiologist detection of malignant lung nodules on chest radiographs. Radiology. 2020;294(1):199–209.
Yoo H, Kim KH, Singh R, Digumarthy SR, Kalra MK. Validation of a deep learning algorithm for the detection of malignant pulmonary nodules in chest radiographs. JAMA Netw Open. 2020;3(9): e2017135.
Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, Fagerstrom RM, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365(5):395–409.
Lu MT, Raghu VK, Mayrhofer T, Aerts H, Hoffmann U. Deep learning using chest radiographs to identify high-risk smokers for lung cancer screening computed tomography: development and validation of a prediction model. Ann Intern Med. 2020;173(9):704–13.
Raghu VK, Walia AS, Zinzuwadia AN, Goiffon RJ, Shepard JO, Aerts H, et al. Validation of a deep learning-based model to predict lung cancer risk using chest radiographs and electronic medical record data. JAMA Netw Open. 2022;5(12): e2248793.
Huang P, Lin CT, Li Y, Tammemagi MC, Brock MV, Atkar-Khattra S, et al. Prediction of lung cancer risk at follow-up screening with low-dose CT: a training and validation study of a deep learning method. The Lancet Digital health. 2019;1(7):e353–62.
Venkadesh KV, Setio AAA, Schreuder A, Scholten ET, Chung K, Wile MMW, et al. Deep learning for malignancy risk estimation of pulmonary nodules detected at low-dose screening CT. Radiology. 2021;300(2):438–47.
Mikhael PG, Wohlwend J, Yala A, Karstens L, **ang J, Takigami AK, et al. Sybil: a validated deep learning model to predict future lung cancer risk from a single low-dose chest computed tomography. J Clin Oncol: Off J Am Soci Clin Oncol. 2023;41(12):2191–200.
Yi L, Zhang L, Xu X, Guo J. Multi-label softmax networks for pulmonary nodule classification using unbalanced and dependent categories. IEEE Trans Med Imaging. 2023;42(1):317–28.
Luo X, Song T, Wang G, Chen J, Chen Y, Li K, et al. SCPM-Net: an anchor-free 3D lung nodule detection network using sphere representation and center points matching. Med Image Anal. 2022;75: 102287.
Wang G, Qiu M, **ng X, Zhou J, Yao H, Li M, et al. Lung cancer scRNA-seq and lipidomics reveal aberrant lipid metabolism for early-stage diagnosis. Sci Transl Med. 2022;14(630):eabk2756.
Hollon T, Jiang C, Chowdury A, Nasir-Moin M, Kondepudi A, Aabedi A, et al. Artificial-intelligence-based molecular classification of diffuse gliomas using rapid, label-free optical imaging. Nat Med. 2023;29(4):828–32.
Corley DA, Jensen CD, Marks AR, Zhao WK, Lee JK, Doubeni CA, et al. Adenoma detection rate and risk of colorectal cancer and death. N Engl J Med. 2014;370(14):1298–306.
Sinonquel P, Eelbode T, Hassan C, Antonelli G, Filosofi F, Neumann H, et al. Real-time unblinding for validation of a new CADe tool for colorectal polyp detection. Gut. 2021;70(4):641–3.
Wesp P, Grosu S, Graser A, Maurus S, Schulz C, Knösel T, et al. Deep learning in CT colonography: differentiating premalignant from benign colorectal polyps. Eur Radiol. 2022;32(7):4749–59.
Grosu S, Wesp P, Graser A, Maurus S, Schulz C, Knösel T, et al. Machine learning-based differentiation of benign and premalignant colorectal polyps detected with CT colonography in an asymptomatic screening population: a proof-of-concept study. Radiology. 2021;299(2):326–35.
Troya J, Krenzer A, Flisikowski K, Sudarevic B, Banck M, Hann A, et al. New concept for colonoscopy including side optics and artificial intelligence. Gastrointest Endosc. 2022;95(4):794–8.
Zhang JK, Fanous M, Sobh N, Kajdacsy-Balla A, Popescu G. Automatic colorectal cancer screening using deep learning in spatial light interference microscopy data. Cells. 2022;11(4):716.
Xu H, Tang RSY, Lam TYT, Zhao G, Lau JYW, Liu Y, et al. Artificial intelligence-assisted colonoscopy for colorectal cancer screening: a multicenter randomized controlled trial. Clin Gastroenterol Hepatol: Off Clin Pract J Am Gastroenterol Assoc. 2023;21(2):337-46.e3.
Kudo SE, Ichimasa K, Villard B, Mori Y, Misawa M, Saito S, et al. Artificial intelligence system to determine risk of t1 colorectal cancer metastasis to lymph node. Gastroenterology. 2021;160(4):1075-84.e2.
Areia M, Mori Y, Correale L, Repici A, Bretthauer M, Sharma P, et al. Cost-effectiveness of artificial intelligence for screening colonoscopy: a modelling study. The Lancet Digital health. 2022;4(6):e436–44.
Hassan C, Balsamo G, Lorenzetti R, Zullo A, Antonelli G. Artificial intelligence allows leaving-in-situ colorectal polyps. Clin Gastroenterol Hepatol: Off Clin Pract J Am Gastroenterol Assoc. 2022;20(11):2505-13.e4.
Soares F, Becker K, Anzanello MJ. A hierarchical classifier based on human blood plasma fluorescence for non-invasive colorectal cancer screening. Artif Intell Med. 2017;82:1–10.
Konishi Y, Okumura S, Matsumoto T, Itatani Y, Nishiyama T, Okazaki Y, et al. Development and evaluation of a colorectal cancer screening method using machine learning-based gut microbiota analysis. Cancer Med. 2022;11(16):3194–206.
Ji M, Zhong J, Xue R, Su W, Kong Y, Fei Y, et al. Early detection of cervical cancer by fluorescence lifetime imaging microscopy combined with unsupervised machine learning. Int J Mol Sci. 2022;23(19):11476.
Wang S, Yin Y, Wang D, Wang Y, ** Y. Interpretability-based multimodal convolutional neural networks for skin lesion diagnosis. IEEE Trans Cybernet. 2022;52(12):12623–37.
Sangers TE, Wakkee M, Kramer-Noels EC, Nijsten T, Lugtenberg M. Views on mobile health apps for skin cancer screening in the general population: an in-depth qualitative exploration of perceived barriers and facilitators. Br J Dermatol. 2021;185(5):961–9.
Alhazmi A, Alhazmi Y, Makrami A, Masmali A, Salawi N, Masmali K, et al. Application of artificial intelligence and machine learning for prediction of oral cancer risk. J Oral Pathol Med: Off Publ Int Assoc Oral Pathol Am Acad Oral Pathol. 2021;50(5):444–50.
Adeoye J, Zheng LW, Thomson P, Choi SW, Su YX. Explainable ensemble learning model improves identification of candidates for oral cancer screening. Oral Oncol. 2023;136: 106278.
Gao Y, **n L, Lin H, Yao B, Zhang T, Zhou AJ, et al. Machine learning-based automated sponge cytology for screening of oesophageal squamous cell carcinoma and adenocarcinoma of the oesophagogastric junction: a nationwide, multicohort, prospective study. Lancet Gastroenterol Hepatol. 2023;8(5):432–45.
Raab SS, Grzybicki DM. Quality in cancer diagnosis. CA: Cancer J Clin. 2010;60(3):139–65.
Veta M, van Diest PJ, Kornegoor R, Huisman A, Viergever MA, Pluim JP. Automatic nuclei segmentation in H&E stained breast cancer histopathology images. PLoS ONE. 2013;8(7): e70221.
Rezaeilouyeh H, Mahoor MH, Zhang JJ, La Rosa FG, Chang S, Werahera PN. Diagnosis of prostatic carcinoma on multiparametric magnetic resonance imaging using shearlet transform. Annual International Conference of the IEEE Engineering in Medicine and Biology Society IEEE Engineering in Medicine and Biology Society Annual International Conference. 2014;2014:6442-5
Kim I, Kang K, Song Y, Kim TJ. Application of artificial intelligence in pathology: trends and challenges. Diagnostics (Basel, Switzerland). 2022;12(11):2794.
Lu MY, Chen TY, Williamson DFK, Zhao M, Shady M, Lipkova J, et al. AI-based pathology predicts origins for cancers of unknown primary. Nature. 2021;594(7861):106–10.
Chen C, Lu MY, Williamson DFK, Chen TY, Schaumberg AJ, Mahmood F. Fast and scalable search of whole-slide images via self-supervised deep learning. Nature Biomed Eng. 2022;6(12):1420–34.
Liu P, Ji L, Ye F, Fu B. AdvMIL: adversarial multiple instance learning for the survival analysis on whole-slide images. Med Image Anal. 2023;91: 103020.
Azevedo Tosta TA, de Faria PR, Neves LA, do Nascimento MZ. Computational normalization of H&E-stained histological images: Progress, challenges and future potential. Artif Intell Med. 2019;95:118–32.
Rana A, Lowe A, Lithgow M, Horback K, Janovitz T, Da Silva A, et al. Use of deep learning to develop and analyze computational hematoxylin and eosin staining of prostate core biopsy images for tumor diagnosis. JAMA Netw Open. 2020;3(5): e205111.
Huang B, Tian S, Zhan N, Ma J, Huang Z, Zhang C, et al. Accurate diagnosis and prognosis prediction of gastric cancer using deep learning on digital pathological images: a retrospective multicentre study. EBioMedicine. 2021;73: 103631.
Shihabuddin AR, Beevi S. Multi CNN based automatic detection of mitotic nuclei in breast histopathological images. Comput Biol Med. 2023;158: 106815.
Schneider G, Schmidt-Supprian M, Rad R, Saur D. Tissue-specific tumorigenesis: context matters. Nat Rev Cancer. 2017;17(4):239–53.
Chang X, Wang J, Zhang G, Yang M, ** Y, ** C, et al. Predicting colorectal cancer microsatellite instability with a self-attention-enabled convolutional neural network. Cell reports Medicine. 2023;4(2): 100914.
Bilal M, Raza SEA, Azam A, Graham S, Ilyas M, Cree IA, et al. Development and validation of a weakly supervised deep learning framework to predict the status of molecular pathways and key mutations in colorectal cancer from routine histology images: a retrospective study. The Lancet Digital health. 2021;3(12):e763–72.
Gerwert K, Schörner S, Großerueschkamp F, Kraeft AL, Schuhmacher D, Sternemann C, et al. Fast and label-free automated detection of microsatellite status in early colon cancer using artificial intelligence integrated infrared imaging. European J Canc (Oxford, England: 1990). 2023;182:122–31.
Blessin NC, Yang C, Mandelkow T, Raedler JB, Li W, Bady E, et al. Automated Ki-67 labeling index assessment in prostate cancer using artificial intelligence and multiplex fluorescence immunohistochemistry. J Pathol. 2023;260(1):5–16.
Wang CW, Muzakky H, Lee YC, Lin YJ. Chao TK 2023 annotation-free deep learning-based prediction of thyroid molecular cancer biomarker BRAF (V600E) from cytological slides. Int J Mol Sci. 2023;24(3):2521.
Abele N, Tiemann K, Krech T, Wellmann A, Schaaf C, Länger F, et al. Noninferiority of artificial intelligence-assisted analysis of ki-67 and estrogen/progesterone receptor in breast cancer routine diagnostics. Modern Pathol: Off J United States Canad Acad Pathol. 2023;36(3): 100033.
Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, et al. Dermatologist-level classification of skin cancer with deep neural networks. Nature. 2017;542(7639):115–8.
Khan MS, Alam KN, Dhruba AR, Zunair H, Mohammed N. Knowledge distillation approach towards melanoma detection. Comput Biol Med. 2022;146: 105581.
Adepu AK, Sahayam S, Jayaraman U, Arramraju R. Melanoma classification from dermatoscopy images using knowledge distillation for highly imbalanced data. Comput Biol Med. 2023;154: 106571.
Wang Y, Wang Y, Cai J, Lee TK, Miao C, Wang ZJ. SSD-KD: a self-supervised diverse knowledge distillation method for lightweight skin lesion classification using dermoscopic images. Med Image Anal. 2023;84: 102693.
Marchetti MA, Nazir ZH, Nanda JK, Dusza SW, D’Alessandro BM, DeFazio J, et al. 3D Whole-body skin imaging for automated melanoma detection. J Eur Acad Dermatol Venereol: JEADV. 2023;37(5):945–50.
Ahmedt-Aristizabal D, Nguyen C, Tychsen-Smith L, Stacey A, Li S, Pathikulangara J, et al. Monitoring of pigmented skin lesions using 3D whole body imaging. Comput Meth Progr Biomed. 2023;232: 107451.
Tajerian A, Kazemian M, Tajerian M, Akhavan MA. Design and validation of a new machine-learning-based diagnostic tool for the differentiation of dermatoscopic skin cancer images. PLoS ONE. 2023;18(4): e0284437.
Venugopal V, Joseph J, Vipin Das M, Kumar NM. An EfficientNet-based modified sigmoid transform for enhancing dermatological macro-images of melanoma and nevi skin lesions. Comput Methods Programs Biomed. 2022;222: 106935.
Ain QU, Al-Sahaf H, Xue B, Zhang M. Automatically diagnosing skin cancers from multimodality images using two-stage genetic programming. IEEE Trans Cybernet. 2023;53(5):2727–40.
Kränke T, Tripolt-Droschl K, Röd L, Hofmann-Wellenhof R, Koppitz M, Tripolt M. New AI-algorithms on smartphones to detect skin cancer in a clinical setting-A validation study. PLoS ONE. 2023;18(2): e0280670.
Freeman K, Dinnes J, Chuchu N, Takwoingi Y, Bayliss SE, Matin RN, et al. Algorithm based smartphone apps to assess risk of skin cancer in adults: systematic review of diagnostic accuracy studies. BMJ (Clinical research ed). 2020;368: m127.
Yi Z, Hu S, Lin X, Zou Q, Zou M, Zhang Z, et al. Machine learning-based prediction of invisible intraprostatic prostate cancer lesions on (68) Ga-PSMA-11 PET/CT in patients with primary prostate cancer. Eur J Nucl Med Mol Imaging. 2022;49(5):1523–34.
Gao P, Shan W, Guo Y, Wang Y, Sun R, Cai J, et al. Development and validation of a deep learning model for brain tumor diagnosis and classification using magnetic resonance imaging. JAMA Netw Open. 2022;5(8): e2225608.
Knabe M, Welsch L, Blasberg T, Müller E, Heilani M, Bergen C, et al. Artificial intelligence-assisted staging in Barrett’s carcinoma. Endoscopy. 2022;54(12):1191–7.
Liang S, Dong X, Yang K, Chu Z, Tang F, Ye F, et al. A multi-perspective information aggregation network for automatedT-staging detection of nasopharyngeal carcinoma. Phys Med Biol. 2022;67(24): 245007.
Demirjian NL, Varghese BA, Cen SY, Hwang DH, Aron M, Siddiqui I, et al. CT-based radiomics stratification of tumor grade and TNM stage of clear cell renal cell carcinoma. Eur Radiol. 2022;32(4):2552–63.
van der Voort SR, Incekara F, Wijnenga MMJ, Kapsas G, Gahrmann R, Schouten JW, et al. Combined molecular subty**, grading, and segmentation of glioma using multi-task deep learning. Neuro Oncol. 2023;25(2):279–89.
Xu Y, Klyuzhin I, Harsini S, Ortiz A, Zhang S, Bénard F, et al. Automatic segmentation of prostate cancer metastases in PSMA PET/CT images using deep neural networks with weighted batch-wise dice loss. Comput Biol Med. 2023;158: 106882.
Wang R, Gu Y, Zhang T, Yang J. Fast cancer metastasis location based on dual magnification hard example mining network in whole-slide images. Comput Biol Med. 2023;158: 106880.
Lin H, Chen H, Graham S, Dou Q, Rajpoot N, Heng PA. Fast ScanNet: fast and dense analysis of multi-gigapixel whole-slide images for cancer metastasis detection. IEEE Trans Med Imaging. 2019;38(8):1948–58.
Ehteshami Bejnordi B, Veta M, van Diest PJ, van Ginneken B, Karssemeijer N, Litjens G, et al. Diagnostic assessment of deep learning algorithms for detection of lymph node metastases in women with breast cancer. JAMA. 2017;318(22):2199–210.
Zhao J, Wang H, Zhang Y, Wang R, Liu Q, Li J, et al. Deep learning radiomics model related with genomics phenotypes for lymph node metastasis prediction in colorectal cancer. Radiotherapy Oncol: J Eur Soci Therapeut Radiol Oncol. 2022;167:195–202.
Wu S, Hong G, Xu A, Zeng H, Chen X, Wang Y, et al. Artificial intelligence-based model for lymph node metastases detection on whole slide images in bladder cancer: a retrospective, multicentre, diagnostic study. Lancet Oncol. 2023;24(4):360–70.
Murai H, Kodama T, Maesaka K, Tange S, Motooka D, Suzuki Y, et al. Multiomics identifies the link between intratumor steatosis and the exhausted tumor immune microenvironment in hepatocellular carcinoma. Hepatology (Baltimore, MD). 2023;77(1):77–91.
Sammut SJ, Crispin-Ortuzar M, Chin SF, Provenzano E, Bardwell HA, Ma W, et al. Multi-omic machine learning predictor of breast cancer therapy response. Nature. 2022;601(7894):623–9.
He X, Liu X, Zuo F, Shi H, **g J. Artificial intelligence-based multi-omics analysis fuels cancer precision medicine. Semin Cancer Biol. 2023;88:187–200.
Srivastava R. Applications of artificial intelligence multiomics in precision oncology. J Cancer Res Clin Oncol. 2023;149(1):503–10.
Zafari N, Bathaei P, Velayati M, Khojasteh-Leylakoohi F, Khazaei M, Fiuji H, et al. Integrated analysis of multi-omics data for the discovery of biomarkers and therapeutic targets for colorectal cancer. Comput Biol Med. 2023;155: 106639.
Stamatoyannopoulos JA. What does our genome encode? Genome Res. 2012;22(9):1602–11.
Saravanan KA, Panigrahi M, Kumar H, Rajawat D, Nayak SS, Bhushan B, et al. Role of genomics in combating COVID-19 pandemic. Gene. 2022;823: 146387.
Chen HZ, Bonneville R, Roychowdhury S. Implementing precision cancer medicine in the genomic era. Semin Cancer Biol. 2019;55:16–27.
Qiu YL, Zheng H, Devos A, Selby H, Gevaert O. A meta-learning approach for genomic survival analysis. Nat Commun. 2020;11(1):6350.
Sahraeian SME, Fang LT, Karagiannis K, Moos M, Smith S, Santana-Quintero L, et al. Achieving robust somatic mutation detection with deep learning models derived from reference data sets of a cancer sample. Genome Biol. 2022;23(1):12.
Sun JX, He Y, Sanford E, Montesion M, Frampton GM, Vignot S, et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Computat Biol. 2018;14(2):e1005965.
Gupta P, **dal A, Ahuja G, Sengupta D. A new deep learning technique reveals the exclusive functional contributions of individual cancer mutations. J Biol Chem. 2022;298(8):102177.
Sengupta A, Naresh G, Mishra A, Parashar D, Narad P. Proteome analysis using machine learning approaches and its applications to diseases. Adv Protein Chem Struct Biol. 2021;127:161–216.
Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell. 2018;33(4):721-35.e8.
Singh MP, Rai S, Pandey A, Singh NK, Srivastava S. Molecular subtypes of colorectal cancer: an emerging therapeutic opportunity for personalized medicine. Genes & diseases. 2021;8(2):133–45.
Moreno V, Sanz-Pamplona R. Altered pathways and colorectal cancer prognosis. BMC Med. 2015;13:76.
Ding K, Zhou M, Wang H, Zhang S, Metaxas DN. Spatially aware graph neural networks and cross-level molecular profile prediction in colon cancer histopathology: a retrospective multi-cohort study. The Lancet Digital health. 2022;4(11):e787–95.
Li N, Meng G, Yang C, Li H, Liu L, Wu Y, et al. Changes in epigenetic information during the occurrence and development of gastric cancer. Int J Biochem Cell Biol. 2022;153: 106315.
Zhou X, Chai H, Zhao H, Luo CH, Yang Y. Imputing missing RNA-sequencing data from DNA methylation by using a transfer learning-based neural network. GigaScience. 2020;9(7):giaa076.
Huang Z, Wang J, Yan Z, Guo M. Differentially expressed genes prediction by multiple self-attention on epigenetics data. Brief Bioinform. 2022;23(3):bbac117.
Tsimberidou AM, Fountzilas E, Bleris L, Kurzrock R. Transcriptomics and solid tumors: the next frontier in precision cancer medicine. Semin Cancer Biol. 2022;84:50–9.
Jha A, Quesnel-Vallières M, Wang D, Thomas-Tikhonenko A, Lynch KW, Barash Y. Identifying common transcriptome signatures of cancer by interpreting deep learning models. Genome Biol. 2022;23(1):117.
Weitz P, Wang Y, Kartasalo K, Egevad L, Lindberg J, Grönberg H, et al. Transcriptome-wide prediction of prostate cancer gene expression from histopathology images using co-expression-based convolutional neural networks. Bioinformatics (Oxford, England). 2022;38(13):3462–9.
He B, Bergenstråhle L, Stenbeck L, Abid A, Andersson A, Borg Å, et al. Integrating spatial gene expression and breast tumour morphology via deep learning. Nat Biomed Eng. 2020;4(8):827–34.
Schmauch B, Romagnoni A, Pronier E, Saillard C, Maillé P, Calderaro J, et al. A deep learning model to predict RNA-Seq expression of tumours from whole slide images. Nat Commun. 2020;11(1):3877.
Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19(11):1423–37.
Ståhl PL, Salmén F, Vickovic S, Lundmark A, Navarro JF, Magnusson J, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (New York, NY). 2016;353(6294):78–82.
Lewis SM, Asselin-Labat ML, Nguyen Q, Berthelet J, Tan X, Wimmer VC, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods. 2021;18(9):997–1012.
Zaitsev A, Chelushkin M, Dyikanov D, Cheremushkin I, Shpak B, Nomie K, et al. Precise reconstruction of the TME using bulk RNA-seq and a machine learning algorithm trained on artificial transcriptomes. Cancer Cell. 2022;40(8):879-94.e16.
Bergenstråhle L, He B, Bergenstråhle J, Abalo X, Mirzazadeh R, Thrane K, et al. Super-resolved spatial transcriptomics by deep data fusion. Nat Biotechnol. 2022;40(4):476–9.
Hu J, Coleman K, Zhang D, Lee EB, Kadara H, Wang L, et al. Deciphering tumor ecosystems at super resolution from spatial transcriptomics with TESLA. Cell Syst. 2023;14(5):404-17.e4.
Zhang H, Zhang N, Wu W, Zhou R, Li S, Wang Z, et al. Machine learning-based tumor-infiltrating immune cell-associated lncRNAs for predicting prognosis and immunotherapy response in patients with glioblastoma. Brief Bioinform. 2022;23(6):bbac386.
Zhou M, Zhang Z, Bao S, Hou P, Yan C, Su J, et al. Computational recognition of lncRNA signature of tumor-infiltrating B lymphocytes with potential implications in prognosis and immunotherapy of bladder cancer. Briefings Bioinform. 2021;22(3):bbaa047.
Zhang N, Zhang H, Wu W, Zhou R, Li S, Wang Z, et al. Machine learning-based identification of tumor-infiltrating immune cell-associated lncRNAs for improving outcomes and immunotherapy responses in patients with low-grade glioma. Theranostics. 2022;12(13):5931–48.
Korfiati A, Grafanaki K, Kyriakopoulos GC, Skeparnias I, Georgiou S, Sakellaropoulos G, et al. Revisiting miRNA association with melanoma recurrence and metastasis from a machine learning point of view. Int J Mol Sci. 2022;23(3):1299.
Hosseiniyan Khatibi SM, Ardalan M, Teshnehlab M, Vahed SZ, Pirmoradi S. Panels of mRNAs and miRNAs for decoding molecular mechanisms of Renal Cell Carcinoma (RCC) subtypes utilizing Artificial Intelligence approaches. Sci Rep. 2022;12(1):16393.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32.
Keyl P, Bischoff P, Dernbach G, Bockmayr M, Fritz R, Horst D, et al. Single-cell gene regulatory network prediction by explainable AI. Nucleic Acids Res. 2023;51(4): e20.
Ogunleye AZ, Piyawajanusorn C, Gonçalves A, Ghislat G, Ballester PJ. Interpretable Machine Learning Models to Predict the Resistance of Breast Cancer Patients to Doxorubicin from Their microRNA Profiles. Adv Sci (Weinheim, Baden-Wurttemberg, Germany). 2022;9(24):e2201501.
Wang S, Zhu H, Zhou H, Cheng J, Yang H. MSpectraAI: a powerful platform for deciphering proteome profiling of multi-tumor mass spectrometry data by using deep neural networks. BMC Bioinformatics. 2020;21(1):439.
Dong H, Liu Y, Zeng WF, Shu K, Zhu Y, Chang C. A deep learning-based tumor classifier directly using MS raw data. Proteomics. 2020;20(21–22): e1900344.
Ludwig C, Gillet L, Rosenberger G, Amon S, Collins BC, Aebersold R. Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Mol Syst Biol. 2018;14(8): e8126.
López-Sánchez LM, Jiménez-Izquierdo R, Peñarando J, Mena R, Guil-Luna S, Toledano M, et al. SWATH-based proteomics reveals processes associated with immune evasion and metastasis in poor prognosis colorectal tumours. J Cell Mol Med. 2019;23(12):8219–32.
Nwaokorie A, Fey D. Personalised medicine for colorectal cancer using mechanism-based machine learning models. Int J Mol Sci. 2021;22(18):9970.
Franciosa G, Smits JGA, Minuzzo S, Martinez-Val A, Indraccolo S, Olsen JV. Proteomics of resistance to Notch1 inhibition in acute lymphoblastic leukemia reveals targetable kinase signatures. Nat Commun. 2021;12(1):2507.
Ye X, Yang Y, Zhou J, Xu L, Wu L, Huang P, et al. Combinatory strategy using nanoscale proteomics and machine learning for T cell subty** in peripheral blood of single multiple myeloma patients. Anal Chim Acta. 2021;1173: 338672.
Liang CA, Chen L, Wahed A, Nguyen AND. Proteomics analysis of FLT3-ITD mutation in acute myeloid leukemia using deep learning neural network. Ann Clin Lab Sci. 2019;49(1):119–26.
Kim H, Kim Y, Han B, Jang JY, Kim Y. Clinically applicable deep learning algorithm using quantitative proteomic data. J Proteome Res. 2019;18(8):3195–202.
Deeb SJ, Tyanova S, Hummel M, Schmidt-Supprian M, Cox J, Mann M. Machine learning-based classification of diffuse large B-cell lymphoma patients by their protein expression profiles. Mol Cell Prot: MCP. 2015;14(11):2947–60.
Wishart DS. Metabolomics for investigating physiological and pathophysiological processes. Physiol Rev. 2019;99(4):1819–75.
DePeaux K, Delgoffe GM. Metabolic barriers to cancer immunotherapy. Nat Rev Immunol. 2021;21(12):785–97.
Agarwala PK, Aneja R, Kapoor S. Lipidomic landscape in cancer: actionable insights for membrane-based therapy and diagnoses. Med Res Rev. 2022;42(2):983–1018.
Rodrigues J, Amin A, Raghushaker CR, Chandra S, Joshi MB, Prasad K, et al. Exploring photoacoustic spectroscopy-based machine learning together with metabolomics to assess breast tumor progression in a xenograft model ex vivo. Laboratory Invest: J Tech Meth Pathol. 2021;101(7):952–65.
Murata T, Yanagisawa T, Kurihara T, Kaneko M, Ota S, Enomoto A, et al. Salivary metabolomics with alternative decision tree-based machine learning methods for breast cancer discrimination. Breast Cancer Res Treat. 2019;177(3):591–601.
Ishii H, Saitoh M, Sakamoto K, Sakamoto K, Saigusa D, Kasai H, et al. Lipidome-based rapid diagnosis with machine learning for detection of TGF-β signalling activated area in head and neck cancer. Br J Cancer. 2020;122(7):995–1004.
Tian M, Lin Z, Wang X, Yang J, Zhao W, Lu H, et al. Pure ion chromatograms combined with advanced machine learning methods improve accuracy of discriminant models in LC-MS-based untargeted metabolomics. Molecules (Basel, Switzerland). 2021;26(9):2715.
Ma Y, Zhang P, Wang F, Liu W, Yang J, Qin H. An integrated proteomics and metabolomics approach for defining oncofetal biomarkers in the colorectal cancer. Ann Surg. 2012;255(4):720–30.
Zhou J, Ji N, Wang G, Zhang Y, Song H, Yuan Y, et al. Metabolic detection of malignant brain gliomas through plasma lipidomic analysis and support vector machine-based machine learning. EBioMedicine. 2022;81: 104097.
Yuan Y, Zhao Z, Xue L, Wang G, Song H, Pang R, et al. Identification of diagnostic markers and lipid dysregulation in oesophageal squamous cell carcinoma through lipidomic analysis and machine learning. Br J Cancer. 2021;125(3):351–7.
Wang H, Yin Y, Zhu ZJ. Encoding LC-MS-based untargeted metabolomics data into images toward AI-based clinical diagnosis. Anal Chem. 2023;95(16):6533–41.
Huang L, Wang L, Hu X, Chen S, Tao Y, Su H, et al. Machine learning of serum metabolic patterns encodes early-stage lung adenocarcinoma. Nat Commun. 2020;11(1):3556.
Manzi M, Palazzo M, Knott ME, Beauseroy P, Yankilevich P, Giménez MI, et al. Coupled mass-spectrometry-based lipidomics machine learning approach for early detection of clear cell renal cell carcinoma. J Proteome Res. 2021;20(1):841–57.
Wallace PW, Conrad C, Brückmann S, Pang Y, Caleiras E, Murakami M, et al. Metabolomics, machine learning and immunohistochemistry to predict succinate dehydrogenase mutational status in phaeochromocytomas and paragangliomas. J Pathol. 2020;251(4):378–87.
Alakwaa FM, Chaudhary K, Garmire LX. Deep learning accurately predicts estrogen receptor status in breast cancer metabolomics data. J Proteome Res. 2018;17(1):337–47.
Yang J, Chen Y, **g Y, Green MR, Han L. Advancing CAR T cell therapy through the use of multidimensional omics data. Nat Rev Clin Oncol. 2023;20(4):211–28.
Choi JM, Chae H. moBRCA-net: a breast cancer subtype classification framework based on multi-omics attention neural networks. BMC Bioinform. 2023;24(1):169.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science (New York, NY). 2013;339(6127):1546–58.
Sanders LM, Chandra R, Zebarjadi N, Beale HC, Lyle AG, Rodriguez A, et al. Machine learning multi-omics analysis reveals cancer driver dysregulation in pan-cancer cell lines compared to primary tumors. Commun Biol. 2022;5(1):1367.
Zhang SW, Xu JY, Zhang T. DGMP: identifying cancer driver genes by jointing DGCN and MLP from multi-omics genomic data. Gen Proteom Bioinform. 2022;20(5):928–38.
Zhao W, Gu X, Chen S, Wu J, Zhou Z. MODIG: integrating multi-omics and multi-dimensional gene network for cancer driver gene identification based on graph attention network model. Bioinformatics (Oxford, England). 2022;38(21):4901–7.
Yang H, Gan L, Chen R, Li D, Zhang J, Wang Z. From multi-omics data to the cancer druggable gene discovery: a novel machine learning-based approach. Brief Bioinform. 2023;24(1):bbca528.
Sharma A, Lysenko A, Boroevich KA, Tsunoda T. DeepInsight-3D architecture for anti-cancer drug response prediction with deep-learning on multi-omics. Sci Rep. 2023;13(1):2483.
Khadirnaikar S, Shukla S, Prasanna SRM. Machine learning based combination of multi-omics data for subgroup identification in non-small cell lung cancer. Sci Rep. 2023;13(1):4636.
Park MK, Lim JM, Jeong J, Jang Y, Lee JW, Lee JC, et al. Deep-learning algorithm and concomitant biomarker identification for NSCLC prediction using multi-omics data integration. Biomolecules. 2022;12(12):1839.
Lococo F, Boldrini L, Diepriye CD, Evangelista J, Nero C, Flamini S, et al. Lung cancer multi-omics digital human avatars for integrating precision medicine into clinical practice: the LANTERN study. BMC Cancer. 2023;23(1):540.
Chen CC, Chu PY, Lin HY. Supervised learning and multi-omics integration reveals clinical significance of inner membrane mitochondrial protein (IMMT) in prognostic prediction, tumor immune microenvironment and precision medicine for kidney renal clear cell carcinoma. Int J Mol Sci. 2023;24(10):8807.
Zhu J, Kong W, Huang L, Bi S, Jiao X, Zhu S. Identification of immunotherapy and chemotherapy-related molecular subtypes in colon cancer by integrated multi-omics data analysis. Front Immunol. 2023;14:1142609.
Li Y, Wu Y, Huang M, Zhang Y, Bai Z. Attention-guided multi-scale learning network for automatic prostate and tumor segmentation on MRI. Comput Biol Med. 2023;165: 107374.
Wang J, Peng Y, **g S, Han L, Li T, Luo J. A deep-learning approach for segmentation of liver tumors in magnetic resonance imaging using UNet+. BMC Cancer. 2023;23(1):1060.
Vermeulen C, Pagès-Gallego M, Kester L, Kranendonk MEG, Wesseling P, Verburg N, et al. Ultra-fast deep-learned CNS tumour classification during surgery. Nature. 2023;622(7984):842–9.
Raju S, Peddireddy Veera VR. Classification of brain tumours from MRI images using deep learning-enabled hybrid optimization algorithm. Network (Bristol, England). 2023;34(4):408–37.
Wong CC, Li W, Chan B, Yu J. Epigenomic biomarkers for prognostication and diagnosis of gastrointestinal cancers. Semin Cancer Biol. 2019;55:90–105.
Huang HH, Liang Y. A novel cox proportional hazards model for high-dimensional genomic data in cancer prognosis. IEEE/ACM Trans Comput Biol Bioinf. 2021;18(5):1821–30.
Tian T, Sun J. Variable selection for nonparametric additive Cox model with interval-censored data. Biometr J Biometrische Zeitschrift. 2023;65(1): e2100310.
Tong R, Zhu Z, Ling J. Comparison of linear and non-linear machine learning models for time-dependent readmission or mortality prediction among hospitalized heart failure patients. Heliyon. 2023;9(5): e16068.
Baralou V, Kalpourtzi N, Touloumi G. Individual risk prediction: comparing random forests with Cox proportional-hazards model by a simulation study. Biometrical J Biometrische Zeitschrift. 2022;65(6):2100380.
Fanizzi A, Pomarico D, Rizzo A, Bove S, Comes MC, Didonna V, et al. Machine learning survival models trained on clinical data to identify high risk patients with hormone responsive HER2 negative breast cancer. Sci Rep. 2023;13(1):8575.
Li X, Zhai Z, Ding W, Chen L, Zhao Y, **ong W, et al. An artificial intelligence model to predict survival and chemotherapy benefits for gastric cancer patients after gastrectomy development and validation in international multicenter cohorts. Int J Surg (London, England). 2022;105: 106889.
Afrash MR, Mirbagheri E, Mashoufi M, Kazemi-Arpanahi H. Optimizing prognostic factors of five-year survival in gastric cancer patients using feature selection techniques with machine learning algorithms: a comparative study. BMC Med Inform Decis Mak. 2023;23(1):54.
Arya N, Saha S, Mathur A, Saha S. Improving the robustness and stability of a machine learning model for breast cancer prognosis through the use of multi-modal classifiers. Sci Rep. 2023;13(1):4079.
Kim Y, Kim KH, Park J, Yoon HI, Sung W. Prognosis prediction for glioblastoma multiforme patients using machine learning approaches: development of the clinically applicable model. Radiother Oncol: J Eur Soci Therap Radiol Oncol. 2023;183: 109617.
Lv W, Zhou Z, Peng J, Peng L, Lin G, Wu H, et al. Functional-structural sub-region graph convolutional network (FSGCN): application to the prognosis of head and neck cancer with PET/CT imaging. Comput Methods Progr Biomed. 2023;230: 107341.
Chen S, **ang J, Wang X, Zhang J, Yang S, Yang W, et al. Deep learning-based pathology signature could reveal lymph node status and act as a novel prognostic marker across multiple cancer types. Br J Cancer. 2023;129(1):46–53.
Lee W, Park HJ, Lee HJ, Jun E, Song KB, Hwang DW, et al. Preoperative data-based deep learning model for predicting postoperative survival in pancreatic cancer patients. Int J Surg (London, England). 2022;105: 106851.
Khazaee Fadafen M, Rezaee K. Ensemble-based multi-tissue classification approach of colorectal cancer histology images using a novel hybrid deep learning framework. Sci Rep. 2023;13(1):8823.
Li C, Liu M, Zhang Y, Wang Y, Li J, Sun S, et al. Novel models by machine learning to predict prognosis of breast cancer brain metastases. J Transl Med. 2023;21(1):404.
Li J, Liang Y, Zhao X, Wu C. Integrating machine learning algorithms to systematically assess reactive oxygen species levels to aid prognosis and novel treatments for triple -negative breast cancer patients. Front Immunol. 2023;14:1196054.
Verghese G, Li M, Liu F, Lohan A, Kurian NC, Meena S, et al. Multiscale deep learning framework captures systemic immune features in lymph nodes predictive of triple negative breast cancer outcome in large-scale studies. J Pathol. 2023;260(4):376–89.
Li J, Qiao H, Wu F, Sun S, Feng C, Li C, et al. A novel hypoxia- and lactate metabolism-related signature to predict prognosis and immunotherapy responses for breast cancer by integrating machine learning and bioinformatic analyses. Front Immunol. 2022;13: 998140.
Wang Y, Acs B, Robertson S, Liu B, Solorzano L, Wählby C, et al. Improved breast cancer histological grading using deep learning. Ann Oncol: Off J Eur Soci Med Oncol. 2022;33(1):89–98.
Ding H, Feng Y, Huang X, Xu J, Zhang T, Liang Y, et al. Deep learning-based classification and spatial prognosis risk score on whole-slide images of lung adenocarcinoma. Histopathology. 2023;83(2):211–28.
She Y, ** Z, Wu J, Deng J, Zhang L, Su H, et al. Development and validation of a deep learning model for non-small cell lung cancer survival. JAMA Netw Open. 2020;3(6): e205842.
Hosny A, Parmar C, Coroller TP, Grossmann P, Zeleznik R, Kumar A, et al. Deep learning for lung cancer prognostication: a retrospective multi-cohort radiomics study. PLoS Med. 2018;15(11): e1002711.
Finn CB, Sharpe JE, Tong JK, Kaufman EJ, Wachtel H, Aarons CB, et al. Development of a machine learning model to identify colorectal cancer stage in medicare claims. JCO Clin Cancer Inform. 2023;7: e2300003.
Kleppe A, Skrede OJ, De Raedt S, Hveem TS, Askautrud HA, Jacobsen JE, et al. A clinical decision support system optimising adjuvant chemotherapy for colorectal cancers by integrating deep learning and pathological staging markers: a development and validation study. Lancet Oncol. 2022;23(9):1221–32.
Bertsimas D, Margonis GA, Sujichantararat S, Boerner T, Ma Y, Wang J, et al. Using artificial intelligence to find the optimal margin width in hepatectomy for colorectal cancer liver metastases. JAMA Surg. 2022;157(8): e221819.
Skrede OJ, De Raedt S, Kleppe A, Hveem TS, Liestøl K, Maddison J, et al. Deep learning for prediction of colorectal cancer outcome: a discovery and validation study. Lancet (London, England). 2020;395(10221):350–60.
Deng S, Ding J, Wang H, Mao G, Sun J, Hu J, et al. Deep learning-based radiomic nomograms for predicting Ki67 expression in prostate cancer. BMC Cancer. 2023;23(1):638.
Saito S, Sakamoto S, Higuchi K, Sato K, Zhao X, Wakai K, et al. Machine-learning predicts time-series prognosis factors in metastatic prostate cancer patients treated with androgen deprivation therapy. Sci Rep. 2023;13(1):6325.
Lee C, Light A, Alaa A, Thurtle D, van der Schaar M, Gnanapragasam VJ. Application of a novel machine learning framework for predicting non-metastatic prostate cancer-specific mortality in men using the Surveillance, Epidemiology, and End Results (SEER) database. Lancet Digital health. 2021;3(3):e158–65.
Nimgaonkar V, Krishna V, Krishna V, Tiu E, Joshi A, Vrabac D, et al. Development of an artificial intelligence-derived histologic signature associated with adjuvant gemcitabine treatment outcomes in pancreatic cancer. Cell Report Med. 2023;4(4): 101013.
Li J, Huang L, Liao C, Liu G, Tian Y, Chen S. Two machine learning-based nomogram to predict risk and prognostic factors for liver metastasis from pancreatic neuroendocrine tumors: a multicenter study. BMC Cancer. 2023;23(1):529.
Aung TN, Shafi S, Wilmott JS, Nourmohammadi S, Vathiotis I, Gavrielatou N, et al. Objective assessment of tumor infiltrating lymphocytes as a prognostic marker in melanoma using machine learning algorithms. EBioMedicine. 2022;82: 104143.
Guan X, Lu N, Zhang J. Computed tomography-based deep learning nomogram can accurately predict lymph node metastasis in gastric cancer. Dig Dis Sci. 2023;68(4):1473–81.
Zhang X, Gleber-Netto FO, Wang S, Martins-Chaves RR, Gomez RS, Vigneswaran N, et al. Deep learning-based pathology image analysis predicts cancer progression risk in patients with oral leukoplakia. Cancer Med. 2023;12(6):7508–18.
Singh T, Malik G, Someshwar S, Le HTT, Polavarapu R, Chavali LN, et al. Machine learning heuristics on gingivobuccal cancer gene datasets reveals key candidate attributes for prognosis. Genes. 2022;13(12):2379.
Cricelli I, Marconi E, Lapi F. Clinical decision support system (CDSS) in primary care: from pragmatic use to the best approach to assess their benefit/risk profile in clinical practice. Curr Med Res Opin. 2022;38(5):827–9.
Yun HJ, Kim HJ, Kim SY, Lee YS, Lim CY, Chang HS, et al. Adequacy and effectiveness of watson for oncology in the treatment of thyroid carcinoma. Front Endocrinol. 2021;12: 585364.
Yu SH, Kim MS, Chung HS, Hwang EC, Jung SI, Kang TW, et al. Early experience with Watson for Oncology: a clinical decision-support system for prostate cancer treatment recommendations. World J Urol. 2021;39(2):407–13.
Liu C, Liu X, Wu F, **e M, Feng Y, Hu C. Using artificial intelligence (watson for oncology) for treatment recommendations amongst chinese patients with lung cancer: feasibility study. J Med Internet Res. 2018;20(9): e11087.
Liu Y, Huo X, Li Q, Li Y, Shen G, Wang M, et al. Watson for oncology decision system for treatment consistency study in breast cancer. Clin Exper Med. 2022;23(5):1649–57.
Somashekhar SP, Sepúlveda MJ, Puglielli S, Norden AD, Shortliffe EH, Rohit Kumar C, et al. Watson for Oncology and breast cancer treatment recommendations: agreement with an expert multidisciplinary tumor board. Ann Oncol: Off J Eur Soci Med Oncol. 2018;29(2):418–23.
Zhang T, Tan T, Wang X, Gao Y, Han L, Balkenende L, et al. RadioLOGIC, a healthcare model for processing electronic health records and decision-making in breast disease. Cell Reports Medicine. 2023;4(8): 101131.
Chen RJ, Lu MY, Williamson DFK, Chen TY, Lipkova J, Noor Z, et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell. 2022;40(8):865-78.e6.
Thirunavukarasu AJ, Ting DSJ, Elangovan K, Gutierrez L, Tan TF, Ting DSW. Large language models in medicine. Nature Med. 2023;29(8):1930–40.
Kehl KL, Xu W, Lepisto E, Elmarakeby H, Hassett MJ, Van Allen EM, et al. Natural language processing to ascertain cancer outcomes from medical oncologist notes. JCO Clin Canc Inform. 2020;4:680–90.
Savova GK, Danciu I, Alamudun F, Miller T, Lin C, Bitterman DS, et al. Use of natural language processing to extract clinical cancer phenotypes from electronic medical records. Can Res. 2019;79(21):5463–70.
Remedios D, Remedios A. Transformers, codes and labels: large language modelling for natural language processing in clinical radiology. Eur Radiol. 2023;33(6):4226–7.
Tan R, Lin Q, Low GH, Lin R, Goh TC, Chang CCE, et al. Inferring cancer disease response from radiology reports using large language models with data augmentation and prompting. J Am Med Inform Assoc: JAMIA. 2023;30(10):1657–64.
Rahsepar AA, Tavakoli N, Kim GHJ, Hassani C, Abtin F, Bedayat A. How AI responds to common lung cancer questions: ChatGPT vs google bard. Radiology. 2023;307(5): e230922.
Yeo YH, Samaan JS, Ng WH, Ting PS, Trivedi H, Vipani A, et al. Assessing the performance of ChatGPT in answering questions regarding cirrhosis and hepatocellular carcinoma. Clin Mol Hepatol. 2023;29(3):721–32.
Zhu L, Mou W, Chen R. Can the ChatGPT and other large language models with internet-connected database solve the questions and concerns of patient with prostate cancer and help democratize medical knowledge? J Transl Med. 2023;21(1):269.
Singhal K, Azizi S, Tu T, Mahdavi SS, Wei J, Chung HW, et al. Large language models encode clinical knowledge. Nature. 2023;620(7972):172–80.
van der Laak J, Litjens G, Ciompi F. Deep learning in histopathology: the path to the clinic. Nat Med. 2021;27(5):775–84.
Kelly CJ, Karthikesalingam A, Suleyman M, Corrado G, King D. Key challenges for delivering clinical impact with artificial intelligence. BMC Med. 2019;17(1):195.
Bae S, Choi H, Lee DS. Discovery of molecular features underlying the morphological landscape by integrating spatial transcriptomic data with deep features of tissue images. Nucleic Acids Res. 2021;49(10): e55.
Weidener L, Fischer M. Teaching AI ethics in medical education: a sco** review of current literature and practices. Perspectives on medical education. 2023;12(1):399–410.
Tian Y, Wang S, **ong J, Bi R, Zhou Z, Bhuiyan MZA. Robust and privacy-preserving decentralized deep federated learning training: focusing on digital healthcare applications. In: IEEE/ACM Transactions on computational biology and bioinformatics. 2023;pp.
Kumar R, Kumar J, Khan AA, Zakria, Ali H, Bernard CM, et al. Blockchain and homomorphic encryption based privacy-preserving model aggregation for medical images. Comput Med Imag Graph: Off J Comput Med Imag Soci. 2022;102:102139.
Ali A, Almaiah MA, Hajjej F, Pasha MF, Fang OH, Khan R, et al. An Industrial IoT-based blockchain-enabled secure searchable encryption approach for healthcare systems using neural network. Sensors (Basel, Switzerland). 2022;22(2):572.
Freeman K, Geppert J, Stinton C, Todkill D, Johnson S, Clarke A, et al. Use of artificial intelligence for image analysis in breast cancer screening programmes: systematic review of test accuracy. BMJ (Clinical research ed). 2021;374: n1872.
Mathios D, Johansen JS, Cristiano S, Medina JE, Phallen J, Larsen KR, et al. Detection and characterization of lung cancer using cell-free DNA fragmentomes. Nat Commun. 2021;12(1):5060.
Acknowledgements
Not applicable.
Funding
This study was jointly supported by the National Natural Science Foundation of China (U21A20374 and 82072698), Shanghai Municipal Science and Technology Major Project (21JC1401500), Scientific Innovation Project of Shanghai Education Committee (2019-01-07-00-07-E00057), and Natural Science Foundation of Shanghai (23ZR1479300).
Author information
Authors and Affiliations
Contributions
CZ and JX collected the related studies and drafted the manuscript. RT, JY and WW participated in the design of the review. XY and SS initiated the study and revised the manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, C., Xu, J., Tang, R. et al. Novel research and future prospects of artificial intelligence in cancer diagnosis and treatment. J Hematol Oncol 16, 114 (2023). https://doi.org/10.1186/s13045-023-01514-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-023-01514-5