
Abstract
Objective
In this study, we aimed to identify differentially expressed heat shock protein (HSP) profiles in the villi and decidua from patients with early missed abortion (EMA).
Methods
By using high-throughput and high-precision parallel reaction monitoring (PRM)-based targeted proteomics techniques, this study examined the abundance of HSPs in the villi and decidua of 10 patients with EMA and 10 controls. Moreover, the abundance of 3 HSPs in the villi of another 22 patients with EMA and 22 controls was verified with Western blotting and immunohistochemistry (IHC).
Results
There were potential differences in the abundance of 16 HSPs and 42 polypeptides in human villi and decidua compared with those of the control group. Among them, HSP90AB1, HSPD1 and HSPA13 were downregulated in abundance in villi of patients with EMA, with a statistically significant difference, which was consistent with the verification results of Western blots and IHC.
Conclusion
Using a PRM-based targeted proteomics technique, this study is the first to screen and quantitatively analyze the expression profile of HSPs in the villi and decidua of patients with EMA. The significant downregulation of HSP90AB1, HSPD1 and HSPA13 was found to have a potentially intimate association with the occurrence of EMA. The findings in our study may provide novel potential research targets related to HSPs for the pathogenesis, prevention and treatment of EMA.
Similar content being viewed by others

Introduction
Early missed abortion (EMA) refers to the phenomenon where an embryo or fetus has died and is retained in the uterine cavity and fails to expel spontaneously during pregnancy ≤ 12 weeks [1]. Although there are many causes of missed abortion, such as maternal endocrine dysfunction, immune dysfunction, reproductive organ anatomical abnormalities and diseases, systemic diseases and infections, chromosomal abnormalities, etc., the etiologies of more than half of cases of missed abortions remain unknown. Therefore, studies on the pathogenesis of missed abortions can pave the way for new research into the prevention and treatment of EMA.
As molecular chaperones, HSPs, which have been widely found at the maternal–embryonic interface, play an important role in protein folding/unfolding, cell cycle regulation and cellular protection [2, 3]. Recently, according to systematic gene symbols, Kam**a et al., divided the human HSP family into the following major classes: HSPA (HSP70), HSPC (HSP90), HSPD/E (HSP60/HSP10) and CCT (TRiC), HSPH (HSP110), DNAJ (HSP40) and HSPB (small HSP) [4]. Accumulating evidence suggests that HSPs promote decidualization, implantation and placentation, with dysregulated expression resulting in implantation failure, pregnancy loss and other maternal–fetal complications [3]. For example, HSP27 promotes decidualization [5], HSP105 facilitates placental implantation [6], and altered abundance of Hsp70 may lead to the occurrence of abortion [7,8,9]. Therefore, we believe that systematic evaluation of HSP abundance in EMA may lead to new molecular targets.
In our study, a PRM-based targeted proteomics technique was used to detect the differential abundance of HSPs at the proteomic level. This method successfully identified the differential abundance of HSPs during malignant melanoma metastasis and revealed that DNAJB4 is a suppressor of melanoma metastasis [10]. Moreover, it has been demonstrated that altered abundance of HSP70, HSP40, and HSPB8 may be associated with radio-resistance development in breast cancer [11]. In this paper, a quantitative analysis was conducted for 42 peptides of 16 target proteins in villi and decidua from 10 patients with EMA and 10 healthy controls to find new molecular targets.
Materials and methods
Patients and controls
A total of 32 pregnant women who underwent complete curettage of the uterine cavity in Jiangxi Maternal and Child Health Hospital from January 2020 to December 2020 were enrolled in the case group, and 32 healthy women who voluntarily requested induced abortion due to unintended pregnancy during the corresponding gestational period were enrolled in the control group. In the diagnosis of missed abortion, transvaginal ultrasonography is primarily relied upon to detect empty gestational sacs or embryos/fetuses without cardiovascular beats. All included pregnant women had been screened for fetal chromosomal abnormalities, endocrine diseases, anatomical abnormalities, infections, immune diseases, trauma and medical diseases. All participants were also within 6–10 weeks of gestation and had no history of adverse pregnancy. The embryonic villous tissue and maternal decidual tissue were collected respectively during vacuum aspiration under intravenous anesthesia. All samples were immediately processed in liquid nitrogen and then stored in a -80 °C refrigerator. Ten villus and decidual samples in the EMA group and the control group were selected for PRM-based targeted proteomic analysis (see Table 1 for clinical baseline characteristics of the two groups). Based on the PRM results, Western blotting and immunohistochemistry were performed on another 22 villus samples from the EMA group and the control group. Compared with those of the control group, the clinical parameters of another 22 cases also showed no significant differences in maternal age, BMI, pregnancy duration, gravidity, parity and induced abortion. This study was supported by the Ethics Committee of Jiangxi Maternal and Child Health Hospital. All the studies provided informed consent.
Protein extraction and enzymatic hydrolysis
After an appropriate amount of SDT lysis solution was added, the sample was transferred to a 2 ml centrifuge tube preloaded with arenaceous quartz and a 1/4-inch ceramic bead MP 6540–424. Homogenization processing was performed using an MP homogenizer (24 × 2, 6.0 M/S, 60 s, twice). In the subsequent steps, the samples underwent an ultrasonic treatment (180 W for 10 s with intervals of 10 s for a total of ten cycles) and were boiled in a water bath for ten minutes. The supernatant was collected by centrifugation at 12,000 × g for 30 min and filtered through a 0.22 µm membrane to obtain the filtrate. Protein quantification was performed using the bicinchoninic acid (BCA) method, and 20 μg of protein from each sample was used for SDS‒PAGE electrophoresis. The areas of degradation were observed as distinct bands against a blue-stained background after Coomassie Brilliant Blue R-250 protein staining. The sample was subpackaged and stored at -80 °C for preservation.
Following enzymatic hydrolysis with FASP, the resulting peptide was subjected to C18 cartridge desalting. The lyophilized peptides were then reconstituted in a 40 µl solution of 0.1% formic acid and quantified using an enzyme-labeled assay.
Mass spectrometry analysis
A peptide mixture from the sample was prepared, and 1 μg was subjected to chromatographic separation using an HPLC system. Buffer solution A consisted of a 0.1% formic acid aqueous solution, while solution B comprised a 0.1% formic acid acetonitrile aqueous solution (84% acetonitrile). The column was equilibrated with 95% solvent A. The samples were separated via a gradient chromatography column. After high-performance liquid chromatography was conducted, a Q-Exactive HF mass spectrometer (Thermo Scientific) was utilized to perform qualitative analysis via mass spectrometry. The analysis duration was 60 min, and the positive ion detection method was utilized. The scanning range of the parent ion was set at 300–1800 m/z. The primary mass spectrometry resolution reached 60,000 atm/z200. The AGC target was set to 3e6, and the primary maximum IT was limited to 50 ms. The collection of peptides' secondary mass spectrometry was conducted using the following methods: 20 MS2 scans were triggered after each full scan, with a resolution of 15,000 at m/z200 and an AGC target of 1e5. The maximum IT for secondary mass spectrometry was set to 50 ms. HCD was used as the MS2 activation type, with an isolation window of 1.6Th and normalized collision energy set to 27.
The original mass spectrum data were obtained using Proteome Discoverer v.2.2, with database search parameters set to trypsin/P enzyme and 0 missed cleavages. Peptides scoring above 40 were considered reliable, and 1–3 unique peptides were selected for each protein.
Based on the results of protein qualitative analysis, trusted peptides suitable for PRM analysis were imported into Xcalibur mass spectrometry software for PRM method setting. The peptide mixture of 10 µg was subjected to quantitative analysis using both "full scan" and "PRM" modes. The chromatographic separation and full scanning conditions were consistent with those described above. Subsequently, the mass spectrum data were analyzed utilizing Skyline software to determine the availability of selected peptides based on their repeatability and stability.
PRM detection analysis
The peptide information suitable for PRM analysis mentioned above was imported into Xcalibur software to set up the PRM method. For detection, approximately 1 µg of peptide was taken from each sample, and 20 fmol standard peptide (PRTC: ELGQSGVDTYLQTK) was added. LC-PRM/MS was used with the aforementioned PRM method to detect the target protein in each sample. The original PRM files were analyzed using Skyline 3.5.0, and after a correction was performed for the internal standard peptide signal, the expression level of the target protein in each sample was determined.
Western blotting validation
Twenty micrograms of the extracted villus protein sample was separated by SDS‒PAGE, transferred to a polyvinylidene fluoride (PVDF) membrane and sealed with 5% defatted milk powder for 1 h. The membrane was incubated with diluted primary antibodies overnight at 4 °C. The membrane was incubated with secondary antibodies for 1 h. The horseradish peroxidase (HRP) signal was detected using hypersensitive enhanced chemical luminescence (ECL) chemical reagent. The target protein bands were analyzed in ImageJ.
Human HSP90AB1 antibody (Boster, Wuhan, China, no. BM4191, 1:2000 dilution), human HSPD1 antibody (Boster, Wuhan, China, no. M01280-3, 1:2000 dilution) and human HSPA13 antibody (Proteintech, Wuhan, China, no. 12667–2-AP, 1:2000 dilution) were used as the primary antibodies for Western blot analysis. The membranes were also incubated with anti-GAPDH antibody (ZSGB-Bio, no. TA-08, 1:10,000 dilution) to verify equal protein loading.
Immunohistochemistry validation
The paraffin-embedded villus tissue sections were dewaxed and dehydrated as per routine practice. Citric acid antigen repair buffer was used for antigen repair. For blocking of endogenous peroxidase, the slices were incubated in 3% hydrogen peroxide (H2O2) at room temperature for 25 min. The tissue sections were sealed with 3% BSA at room temperature for 30 min and then incubated with 50 μl of diluted primary antibodies overnight at 4 °C. The primary antibody was anti-HSP90AB1 (Boster, Wuhan, China, no. BM4191, 1:200 dilution), anti-HSPD1 (Boster, Wuhan, China, no. M012803, 1:200 dilution) and anti-HSPA13 (Proteintech, Wuhan, China, no. 12667–2-AP, 1:200 dilution). The slices were incubated with the secondary antibodies of the corresponding species of the primary antibody at room temperature for 50 min. Finally, fresh DAB display solution was added to the slices, the color development time was controlled under the microscope, and the positive color was brownish yellow. All sections were analyzed through a modified H-score scoring system [(percentage of weak intensity area × 1) + (percentage of moderate intensity area × 2) + (percentage of strong intensity area × 3)] by two pathologists blinded to the clinical and molecular data. The H-score is a value between 0 and 300, and the larger the value is, the stronger the comprehensive positive intensity [12].
Statistical analysis
SPSS 20.0 (SPSS, Inc., Chicago, IL, USA) software was used for statistical analysis, and all data are expressed as the mean ± standard deviation (SD). Student's t test was used for comparison of quantitative data between the two groups, and P < 0.05 was considered statistically significant. Enumeration data is chi-square test. All experiments were repeated three times.
Results
Differentially expressed protein identification
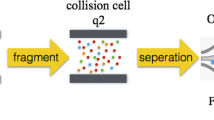
Using PRM-based targeted proteomics, we successfully identified the differentially abundant HSPs in villi and decidua of 10 patients with EMA and 10 controls (Fig. 1). A total of 16 differentially expressed HSPs with 42 polypeptides in total were quantified (Tables 2, 3, 4 and 5). Among them, the abundances of HSP90AB1, HSPD1 and HSPA13 in the villi of the EMA group were decreased compared with those in the control group, with statistically significant differences (all P < 0.05); however, these HSPs were not significantly changed in decidual tissues (P > 0.05).

Workflow of preordered LC-PRM analysis for quantitative evaluation of heat shock proteome in villi and decidua of 10 patients with EMA and 10 Controls. All experiments were performed in triplicate
Western blotting validation of PRM-based results
The Western blotting results showed that the abundance levels of HSP90AB1, HSPD1, and HSPA13 were decreased in 22 cases of EMA villi, with statistically significant differences compared to the control group (Fig. 2). The above results were consistent with those verified by the PRM-based method.

The differential abundance of HSP90AB1, HSPD1 and HSPA13 in villi tissues was verified by Western blot. A The representative of Western blot analysis to verify selected differentially expressed proteins HSP90AB1, HSPD1 and HSPA13 in the villi tissue of embryo from EMA (n = 6) and control(n = 6). B The scatter plot of HSP90AB1 abundance in the villi tissue of embryo from EMA (n = 22) vs control (n = 22) (P < 0.01). C The scatter plot of HSPD1 abundance in the villi tissue of embryo from EMA (n = 22) vs control (n = 22) (P < 0.01). D The scatter plot of HSPA13 abundance in the villi tissue of embryo from EMA (n = 22) vs control (n = 22) (P < 0.01)
Immunohistochemistry validation of PRM-based results
Further analysis was conducted on the 3 candidate biomarkers HSP90AB1, HSPD1 and HSPA13 using IHC. Through staining, it was found that the levels of HSP90AB1, HSPD1 and HSPA13 in the villi of 22 cases of EMA were significantly lower than those in the control group (Fig. 3), which was consistent with the abundance trends detected by the PRM-based method.

A, B, C Immunohistochemical staining for HSP90AB1, HSPD1, and HSPA13 observed in the cytoplasm of syncytiotrophoblast and cytotrophoblast cells (× 200, × 400). Villus from 22 patients with EMA showed lower; D-F Quantitative scoring results of IHC analyses w shown as box plots
Discussion
PRM is a targeted mass spectrometry technique where a new-generation mass spectrometer with high resolution and high precision is used for ion monitoring of the target protein or peptide fragment to realize accurate quantification of the target protein/peptide fragment. In this study, a targeted quantitative proteomics method based on PRM was used to comprehensively analyze the differential abundance of heat shock proteins in EMA tissues. We observed some differences in the abundance of 16 target proteins in human villus and decidual tissues. Specifically, the levels of HSP90AB1, HSPD1 and HSPA13 were decreased in EMA villus tissues with significant differences, which was consistent with the results of Western blot and IHC verification. However, there were no statistically significant changes in these HSPs in decidual tissue, which may be related to the small size of our sample, and these results need to be further confirmed by expanding the sample size; on the other hand, these HSPs may only have changes in expression in villi. Cui et al. [13]. utilized proteomics to investigate factors related to early embryonic development and employed iTRAQ technology to compare protein profiles of serum samples from patients with normal pregnancies and cases of early recurrent spontaneous abortion (ERSA). Through this comparison, they identified 78 differentially expressed proteins. Furthermore, PRM technology was employed to validate three proteins—CD45, PSG1, and Prdx-2—which were found to be closely associated with miscarriage. There are some distinctions between their study and ours. First, we employed distinct clinical samples, focusing on human embryonic chorionic villus and maternal decidua tissues, while they utilized human serum samples. Moreover, we specifically investigated the differential abundance of HSP in the specimens, whereas they examined the differential abundance of all proteins in the specimens. In summary, our findings might provide important evidence for further research into the complex pathophysiological mechanism of EMA.
As a major isoform of the HSP90 family, HSP90AB1 (HSP90β) is usually constitutively expressed [14]. Loones et al. revealed that mouse embryos synthesize high levels of HSP90β during the preimplantation stage of development [15]. Further studies showed that mice with Hsp90β gene mutations failed to differentiate to form the placental labyrinth in the presence of normal Hsp90α abundance [16], suggesting that it plays a critical role in trophoblast differentiation. Moreover, invasion of the decidua by villous trophoblasts plays a crucial role in successful embryo implantation, and the mechanism of embryo implantation is similar to tumor invasion [17]. An increasing number of previous studies have demonstrated that upregulation of HSP90AB1 abundance can promote the invasion and metastasis of various cancers, such as gastric cancer, lung cancer, and colon cancer [18,19,20]. Therefore, it can be assumed that downregulation of HSP90AB1 abundance in aborted villus tissue may lead to reduced differentiation and invasion of trophoblast cells and affect embryo implantation. However, further studies are still needed to explore the mechanisms underlying this reduced abundance.
HSPD1 is constitutively expressed in mitochondria and cytoplasm and plays a key role in chaperoning, thermotolerance, apoptosis, cancer, immunology and embryonic development [21]. HSP60 expression promotes embryonic stem cell (ESC) differentiation and inhibits ESC apoptosis [22]. HSP60 was reported to promote progesterone synthesis [23] and regulate yolk sac erythropoiesis in mice [24]. Lipopolysaccharide-induced implantation failure may be related to significantly lower abundance of Hsp90, Hsp70, and Hsp60 [25]. Cumulatively, these findings suggest that HSP60 plays a distinct role during embryonic development and implantation. In our study, we verified that Hsp60 is obviously downregulated in villi of the EMA group compared with the control group. Similar to our study, using proteomics technology, Johnstone [26] et al. observed that HSP60 was downregulated in highly purified cytotrophoblasts from patients with preeclamptic placentas. Another study showed that the abundance of HSP27, HSP60, HSP70, and HSP90 in syncytiotrophoblasts (STs) and cytotrophoblasts around infarction region placentas with intrauterine fetal growth restriction was decreased due to lethal damage [27]. However, the abundance of HSP60 in peripheral blood was inconsistent with our data, which may be due to the elevation of HSP60 levels in plasma originating from sources other than the trophoblast layer [28]. Further research showed that HSP60 acts as a potent inhibitor of apoptosis by binding to proapoptotic regulators such as Bax, Bak, p21, p53, and survivin [29,30,31]. Gupta et al. observed that HSP60 could form a complex with Bax in the cytosol and inhibit its translocation to mitochondria, leading to the suppression of cell apoptosis [32]. Moreover, apoptosis is increased in EMA villi [33]. Therefore, we speculated that downregulation of HSP60 might promote the apoptosis of villous trophoblast cells and lead to EMA. Further studies will be conducted to verify this hypothesis.
HSPA13 is known as a member of the HSP70 family. To date, research on the biological function of HSPA13 in trophoblasts and its relationship with EMA is still relatively limited. Studies have shown that HSPA13 is overexpressed in colon and hepatocellular carcinoma tissues [34, 35]. Infiltrating gestational trophoblasts share many similarities with tumor cells in biological activity. Therefore, the downregulation of HSPA13 abundance in villus tissue may be critical in the development of EMA disease.
Conclusion
In summary, through PRM-targeted quantitative proteomics, it was found in this study that HSP90AB1, HSPD1 and HSPA13 in villus tissue were candidate proteins with potential importance in EMA. Notably, this method enables more precise and efficient study of changes in protein profiles. However, this study is limited by the small sample size and lack of further functional studies on HSP90AB1, HSPD1 and HSPA13, so the specific mechanisms by which they are involved in the occurrence of EMA have not been clarified. In future studies, we will further investigate the specific functions of these three proteins and their roles in the pathological mechanisms of EMA and identify relevant therapeutic targets. In summary, their potential applications at the maternal–fetal interface would require larger and more in-depth studies.
Availability of data and materials
The data supporting the findings of this study can be obtained from the corresponding author upon reasonable request.
References
Jurkovic D, Overton C, Bender-Atik R. Diagnosis and management of first trimester miscarriage. BMJ. 2013;346:f3676.
De Maio A, Vazquez D. Extracellular heat shock proteins: a new location, a new function. Shock. 2013;40(4):239–46.
Jee B, Dhar R, Singh S, Karmakar S. Heat shock proteins and their role in pregnancy: redefining the function of “old rum in a new bottle.” Front Cell Dev Biol. 2021;9: 648463.
Kam**a HH, Hageman J, Vos MJ, et al. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14(1):105–11.
Ciocca DR, Stati AO, Fanelli MA, Gaestel M. Expression of heat shock protein 25,000 in rat uterus during pregnancy and pseudopregnancy. Biol Reprod. 1996;54(6):1326–35.
Yuan JX, **ao LJ, Lu CL, et al. Increased expression of heat shock protein 105 in rat uterus of early pregnancy and its significance in embryo implantation. Reprod Biol Endocrinol. 2009;7:23.
Gulic T, Laskarin G, Dominovic M, et al. Potential role of heat-shock protein 70 and interleukin-15 in the pathogenesis of threatened spontaneous abortions. Am J Reprod Immunol. 2016;76(2):126–36.
Jain CV, Jessmon P, Barrak CT, et al. Trophoblast survival signaling during human placentation requires HSP70 activation of MMP2-mediated HBEGF shedding. Cell Death Differ. 2017;24(10):1772–83.
Liu M, Sun X, Zhu L, et al. Long noncoding RNA RP11–115N4.1 promotes inflammatory responses by interacting with HNRNPH3 and enhancing the transcription of HSP70 in unexplained recurrent spontaneous abortion. Front Immunol. 2021;12:717785.
Miao W, Li L, Wang Y. A Targeted proteomic approach for heat shock proteins reveals DNAJB4 as a suppressor for melanoma metastasis. Anal Chem. 2018;90(11):6835–42.
Miao W, Fan M, Huang M, et al. Targeted profiling of heat shock proteome in radioresistant breast cancer cells[J]. Chem Res Toxicol. 2019;32(2):326–32.
Paschalis A, Sheehan B, Riisnaes R, et al. Prostate-specific membrane antigen heterogeneity and DNA repair defects in prostate cancer. Eur Urol. 2019;76(4):469–78.
Cui Y, He L, Yang CY, Ye Q. iTRAQ and PRM-based quantitative proteomics in early recurrent spontaneous abortion: biomarkers discovery. Clin Proteomics. 2019;18(16):36.
Sreedhar AS, Kalmár E, Csermely P, Shen YF. Hsp90 isoforms: functions, expression and clinical importance. FEBS Lett. 2004;562(1–3):11–5.
Loones MT, Rallu M, Mezger V, Morange M. HSP gene expression and HSF2 in mouse development. Cell Mol Life Sci. 1997;53(2):179–90.
Voss AK, Thomas T, Gruss P. Mice lacking HSP90beta fail to develop a placental labyrinth. Development. 2000;127(1):1–11.
Abbas Y, Turco MY, Burton GJ, Moffett A. Investigation of human trophoblast invasion in vitro. Hum Reprod Update. 2020;26(4):501–13.
Wang H, Deng G, Ai M, et al. Hsp90ab1stabilizes LRP5 to promote epithelial-mesenchymal transition via activating of AKT and Wnt/β-catenin signaling pathways in gastric cancer progression. Oncogene. 2019;38(9):1489–507.
Biaoxue R, **ling J, Shuanying Y, et al. Upregulation of Hsp90-beta and annexin A1 correlates with poor survival and lymphatic metastasis in lung cancer patients. J Exp Clin Cancer Res. 2012;31(1):70.
de la Mare JA, Jurgens T, Edkins AL. Extracellular Hsp90 and TGFβ regulate adhesion, migration and anchorage independent growth in a paired colon cancer cell line model. BMC Cancer. 2017;17(1):202.
Malik JA, Lone R. Heat shock proteins with an emphasis on HSP 60. Mol Biol Rep. 2021;48(10):6959–69.
Seo NH, Lee EH, Seo JH, et al. HSP60 is required for stemness and proper differentiation of mouse embryonic stem cells. Exp Mol Med. 2018;50(3): e459.
Monreal-Flores J, Espinosa-García MT, García-Regalado A, et al. The heat shock protein 60 promotes progesterone synthesis in mitochondria of JEG-3 cells. Reprod Biol. 2017;17(2):154–61.
Duan Y, Wang H, Mitchell-Silbaugh K, et al. Heat shock protein 60 regulates yolk sac erythropoiesis in mice. Cell Death Dis. 2019;10(10):766.
Jaiswal MK, Agrawal V, Jaiswal YK, et al. Lipopolysaccharide drives alternation of heat shock proteins and induces failure of blastocyst implantation in mouse. Biol Reprod. 2013;88(6):162.
Johnstone ED, Sawicki G, Guilbert L, et al. Differential proteomic analysis of highly purified placental cytotrophoblasts in pre-eclampsia demonstrates a state of increased oxidative stress and reduced cytotrophoblast antioxidant defense. Proteomics. 2011;11(20):4077–84.
Wataba K, Saito T, Takeuchi M, et al. Changed expression of heat shock proteins in various pathological findings in placentas with intrauterine fetal growth restriction. Med Electron Microsc. 2004;37(3):170–6.
Álvarez-Cabrera MC, Barrientos-Galeana E, Barrera-García A, et al. Secretion of heat shock -60, -70 kD protein, IL-1β and TNFα levels in serum of a term normal pregnancy and patients with pre-eclampsia development. J Cell Mol Med. 2018;22(11):5748–52.
Huang YH, Yeh CT. Functional compartmentalization of HSP60-Survivin interaction between mitochondria and cytosol in cancer cells. Cells. 2019;9(1):23.
Gupta S, Knowlton AA. HSP60, Bax, apoptosis and the heart [J]. J Cell Mol Med. 2005;9(1):51–8.
Ghosh JC, Dohi T, Kang BH. Hsp60 regulation of tumor cell apoptosis. J Biol Chem. 2008;283(8):5188–94.
Gupta S, Knowlton AA. Cytosolic heat shock protein 60, hypoxia and apoptosis. Circulation. 2002;106(21):2727–33.
Zhang Y, Zhou J, Li MQ, et al. MicroRNA-184 promotes apoptosis of trophoblast cells via targeting WIG1 and induces early spontaneous abortion. Cell Death Dis. 2019;10(3):223.
Guan Y, Zhu X, Liang J, et al. Upregulation of HSPA1A/HSPA1B/HSPA7 and downregulation of HSPA9 were related to poor survival in colon cancer. Front Oncol. 2021;11: 749673.
Wang B, Lan T, **ao H, et al. The expression profiles and prognostic values of HSP70s in hepatocellular carcinoma. Cancer Cell Int. 2021;21(1):286.
Acknowledgements
We appreciate Shanghai Applied Protein Technology (Shanghai, China) for the technical support of LC–PRM experiment.
Funding
This work was supported by grants from the Natural Science Foundation of Jiangxi Province (No. 20171BAB205013) and Jiangxi Provincial Health and Family Planning Commission science and technology plan (20195472).
Author information
Authors and Affiliations
Contributions
Shu-Hui Huang and Qi Chen conceived and designed the project; **ao-Qing Chen and Hai-Lian Luo performed Western blotting and Immunohistochemistry; Chen **ao-Qing and Li-Na **a collected and analyzed data; **ao-Fang Chen wrote the manuscript; All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was supported by the Ethics Committee of Jiangxi Maternal and Child Health Hospital. All the studies provided informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. Western blot analysis to verify selected differentially expressed proteins HSP90AB1, HSPD1 and HSPA13; Candidate proteins were examined in triplicate and normalized to GAPDH levels for quantitative analysis; 1-6represent sample 1 to sample 6.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, XF., Chen, XQ., Luo, HL. et al. PRM-based quantitative proteomics analysis of altered HSP abundance in villi and decidua of patients with early missed abortion. Proteome Sci 21, 12 (2023). https://doi.org/10.1186/s12953-023-00213-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12953-023-00213-w