
Abstract
Background
Myocarditis substantially increases the risk of ventricular arrhythmia. Approximately 30% of all ventricular arrhythmia cases in patients with myocarditis originate from the right ventricular outflow tract (RVOT). However, the role of NLRP3 signaling in RVOT arrhythmogenesis remains unclear.
Methods
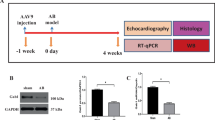
Rats with myosin peptide–induced myocarditis (experimental group) were treated with an NLRP3 inhibitor (MCC950; 10 mg/kg, daily for 14 days) or left untreated. Then, they were subjected to electrocardiography and echocardiography. Ventricular tissue samples were collected from each rat’s RVOT, right ventricular apex (RVA), and left ventricle (LV) and examined through conventional microelectrode and histopathologic analyses. In addition, whole-cell patch-clamp recording, confocal fluorescence microscopy, and Western blotting were performed to evaluate ionic currents, intracellular Ca2+ transients, and Ca2+-modulated protein expression in individual myocytes isolated from the RVOTs.
Results
The LV ejection fraction was lower and premature ventricular contraction frequency was higher in the experimental group than in the control group (rats not exposed to myosin peptide). Myocarditis increased the infiltration of inflammatory cells into cardiac tissue and upregulated the expression of NLRP3; these observations were more prominent in the RVOT and RVA than in the LV. Furthermore, experimental rats treated with MCC950 (treatment group) improved their LV ejection fraction and reduced the frequency of premature ventricular contraction. Histopathological analysis revealed higher incidence of abnormal automaticity and pacing-induced ventricular tachycardia in the RVOTs of the experimental group than in those of the control and treatment groups. However, the incidences of these conditions in the RVA and LV were similar across the groups. The RVOT myocytes of the experimental group exhibited lower Ca2+ levels in the sarcoplasmic reticulum, smaller intracellular Ca2+ transients, lower L-type Ca2+ currents, larger late Na+ currents, larger Na+–Ca2+ exchanger currents, higher reactive oxygen species levels, and higher Ca2+/calmodulin-dependent protein kinase II levels than did those of the control and treatment groups.
Conclusion
Myocarditis may increase the rate of RVOT arrhythmogenesis, possibly through electrical and structural remodeling. These changes may be mitigated by inhibiting NLRP3 signaling.
Similar content being viewed by others

Background
Myocarditis is a nonischemic cardiomyopathy involving the infiltration of cardiac tissue by inflammatory cells and has infectious, noninfectious, autoimmune, and miscellaneous etiologies. This condition often remains undiagnosed; however, it is detected during approximately 9% of all routine postmortem examinations [1,2,3]. Myocarditis has heterogeneous clinical manifestations, ranging from nonspecific symptoms to rapidly declining cardiac function and life-threatening arrhythmia [2]. The prevalence of myocarditis is 9%–40% in patients with idiopathic cardiomyopathy [4, 5] and approximately 50% in individuals with ventricular arrhythmia (VA) [6]. In a cohort study, approximately 51% of all patients experiencing frequent premature ventricular contractions (PVCs) had underlying myocardial inflammation [7]. A considerable proportion (30%) of all ventricular arrhythmia cases in patients with myocarditis originate from the right ventricular outflow tract (RVOT) [8]. Evidence suggests an association between COVID-19 and myocarditis [9, 10]: an increased incidence of myocarditis has been discovered in patients with COVID-19, particularly those with severe or critical disease [11]. This observation has drawn attention to the pathogenesis, clinical presentation, and management of COVID-19-associated myocarditis. Myocytes constituting the RVOT exhibit distinct electrophysiological characteristics associated with a high propensity for arrhythmogenesis [12, 13]. However, whether myocarditis promotes RVOT arrhythmogenesis, and thus increases the risk of VA, remains unclear. Moreover, little is known regarding the mechanisms underlying myocarditis-induced VA.
Myocarditis, characterized by inflammation of the cardiac muscle, can trigger a cascade of immune responses that markedly affect the heart’s function and structure. Central to this immune response is activation of the nucleotide oligomerization domain-like receptor family protein 3 (NLRP3) inflammasome; this phenomenon has been increasingly recognized as a key mediator of the myocardial inflammatory response [26].
ICa-L was measured as an inward current during depolarization from a holding potential of − 50 mV to test potentials ranging from − 40 to + 60 mV in 10‐mV steps over a 300-ms period at a frequency of 0.1 Hz. For this, we used a perforated patch-clamp with amphotericin B (300 μg/mL). NaCl and KCl present in the external solution were replaced by tetraethylammonium chloride and CsCl, respectively. The peak current amplitude was selected to represent ICa-L.
The NCX current was elicited using test pulses ranging from − 100 to + 100 mV from a holding potential of − 40 mV over a 300-ms period at a frequency of 0.1 Hz. Notably, the NCX current was estimated by subtracting the currents sensitive to 10 mM Ni2+ under control conditions from those recorded in the presence of Ni2+. This approach helped measure the exchange of Na+ and Ca2+ ions in terms of the NCX current. The external solution comprised 140 mM NaCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose (pH 7.4), 10 μM strophanthidin, 10 μM nitrendipine, 100 μM niflumic acid, and 5 mM HEPES.
Measurement of intracellular reactive oxygen species and Na+ level
Cross-sectional areas of the isolated RVOT myocytes were visualized using a confocal laser scanning microscope (Zeiss LSM 510; Carl Zeiss). The images were processed using ImageJ [20]. CellROX green (Life Technologies, Grand Island, NY, USA) was used to measure the cytosolic levels of reactive oxygen species (ROS), whereas MitoSOX Red (Life Technologies) was used to measure the mitochondrial levels of ROS. Asante NaTRIUM Green-2 AM (Teflabs, Austin, TX, USA) was employed to measure the cytosolic levels of Na+ in RVOT myocytes freshly isolated from the control, experimental, and treatment groups. Microscopic examinations were performed using the aforementioned Zeiss microscope and an inverted microscope (Axiovert 100) with a 63 × 1.25 numerical aperture oil immersion objective, as described previously [27]. CellROX green, MitoSOX Red, and Asante NaTRIUM Green-2 were excited at 488 nm, and fluorescence signals were acquired at a wavelength of ≥ 505 nm by using the confocal system in the XY mode. During the experiment, RVOT myocytes were paced at a frequency of 1 Hz. Fluorescence images were analyzed using Image-Pro Plus (version 6.0) and SigmaPlot (version 12), as described previously [20].
Western blotting
Changes in the expression of ion channels and Ca2+-modulated proteins were investigated through Western blotting. RVOT tissues were homogenized and centrifuged in buffer systems. Equal amounts of total protein were separated through 5% or 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The resultant protein bands were electrophoretically transferred onto polyvinylidene difluoride membranes. For the immunofluorescence-based detection of gap junction proteins, all blots were stained with primary antibodies against sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a), Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylated at Thr286 (pCaMKII), RyR2, total phospholamban (Thermo Fisher Scientific, Rockford, IL, USA), phosphorylated RyR at Ser2808, phospholamban phosphorylated at Thr17 (Badrilla, UK), catalytic subunit of protein kinase A (BD BioSciences, USA), Cav1.2, NLRP3, nuclear factor (NF)-κB, IL-1β, and glyceraldehyde-3-phosphate dehydrogenase (MBL, Japan); all secondary antibodies were conjugated with horseradish peroxidase. Bound antibodies were detected using an enhanced chemiluminescence detection system and were analyzed using the AlphaEaseFC software. To ensure equal protein loading, all target bands were normalized to a band of glyceraldehyde-3-phosphate dehydrogenase.
Statistical analysis
All continuous variables are expressed as means ± standard deviations. One-way analysis of variance with a post hoc Tukey test was employed to compare variables between the control, myocarditis, and MCC950-treated myocarditis groups. Differences between nonparametric variables were analyzed using the chi-square test with Fisher’s exact correction. P < 0.05 was considered statistically significant.
Results
NLRP3 inhibition mitigates myocardial dysfunction and prevents VA in rats with myocarditis
Our echocardiographic analysis of LV function revealed markedly lower LV ejection fractions, greater LV septal wall thickness, and larger left ventricular internal diameters at end diastole and systole in the experimental group than in the control group (Fig. 1D). However, MCC950 treatment significantly mitigated the dysfunction of the LV and the dilation of the left ventricular internal diameters at end diastole and systole (Fig. 1D). In vivo monitoring of the experimental group indicated abnormal ECG morphologies on day 21 (Fig. 1C), characterized by QRS fragmentation—a typical ECG finding for myocarditis. The experimental group also exhibited longer QT intervals, corrected QT intervals (QTc), and QRS durations than did the control and treatment groups (Fig. 1B). Notably, the three groups had similar RR and PR intervals, suggesting that overall heart rate and atrioventricular conduction were not significantly affected by myocarditis or MCC950 treatment. Furthermore, after receiving an intraperitoneal injection of caffeine (120 mg/kg), the experimental group (n = 6) exhibited a higher rate of VA incidence than did the control (n = 6) and treatment (n = 10) groups; the groups were compared in terms of spontaneous PVCs (50% vs. 0% and 0%, respectively; P < 0.05 for both) and ventricular tachycardia (VT; 20% vs. 0% [P = 0.145] and 0% [P = 0.12], respectively; Fig. 1E,F). These findings suggest that caffeine increases the risk of myocarditis-associated arrhythmogenesis in rats.
Myosin peptide evokes an inflammatory response during early stage myocarditis in rats
Histological analysis revealed the infiltration of immune cells into the cardiac tissue of the experimental group on day 21 (Fig. 2). This infiltration, indicative of myocardial inflammation, was accompanied by localized expansion of the interstitial space due to cardiomyocyte necrosis, a hallmark of the “infarct-like” phenotype associated with myocarditis. Notably, we observed inflammatory cells such as lymphocytes and macrophages within the myocardium. The degree of inflammatory cell infiltration was significantly higher in the experimental group than in the control group; however, this increase was mitigated by MCC950 treatment (Fig. 2A,B). Furthermore, in the experimental group, a more pronounced inflammatory response was observed in the RVOT than in the LV. MCC950 treatment effectively alleviated this inflammatory response. Immunohistochemical staining revealed significantly higher levels of NLRP3 expression in the experimental and MCC950 treatment groups than in the control group. In the experimental group, the level of NLRP3 expression was significantly higher in the RVOT than in the LV (Fig. 2C). This finding suggests variation in NLRP3 expression across cardiac regions in rats with myocarditis.

Results of hematoxylin–eosin and immunohistochemical staining performed to measure NLRP3 expression. Representative sections (day 21) of the RVOTs, RVAs, and LVs of the control (healthy rats), experimental (rats with myocarditis), and treatment (MCC950-treated rats with myocarditis) groups after hematoxylin–eosin staining and immunohistochemical staining for CD45 and NLRP3 (magnification, 400 ×). A Average counts of inflammatory cells (lymphocytes and macrophages) in the control (n = 3), experimental (n = 4), and treatment (n = 3) groups. Macrophages (red arrow) and lymphocytes (yellow arrow) were observed in addition to interstitial space edema (blue arrow) and cardiomyocyte necrosis (green arrow). B Relative expression of CD45 in the RVOTs of the control group was compared with that in different heart regions of the experimental and treatment groups. Average CD45 immunostaining results of the control (n = 4), experimental (n = 4), and treatment (n = 4) groups. C Relative expression of NLRP3 in the RVOTs of the control group was compared with that in the experimental and treatment groups. Average NLRP3 immunostaining results of the control (n = 4), experimental (n = 4), and MCC950-treated (n = 4) groups. RVOT, right ventricle outflow tract; RVA, right ventricular apex; LV, left ventricle
AP morphologies in the RVA, RVOT, and LV of the study groups
The RVOTs of the experimental group exhibited a longer APD90 than did the RVOTs of the control and treatment groups (Fig. 3). However, similar APD50 and APD20 values were found in the three groups. Moreover, no significant between-group difference was observed in the APD20, APD50, or APD90 corresponding to the RVA or LV. The triggered activity rates were higher in the RVOTs of the experimental group than in the RVOTs of the control and treatment groups. However, similar rates of triggered activity in the RVAs and LVs were observed among the groups (Fig. 3). Rapamycin treatment (100 nM) resulted in a 20-beat pause following 2-Hz field stimulation, leading to the induction of VT in the RVOTs of the experimental group but not those of the control group. However, during pacing and after rapamycin treatment, the rate of VT incidence was lower in the RVOTs of the treatment group than in the RVOTs of the experimental group. Notably, the same pause protocol led to a lower rate of VT incidence in the RVAs than in the RVOTs but no VT incidence in the LVs. Thus, the RVOT may be more susceptible to myocarditis-induced arrhythmia than are other ventricular regions. MCC950 treatment appears to effectively mitigate the myocarditis-induced increase in the risk of arrhythmia (Fig. 3).

Morphology of AP and incidence of premature ventricular contractions and ventricular tachycardia. A Superimposed traces depicting APs in the RVOTs of the control (healthy rats), experimental (rats with myocarditis), and treatment (MCC950-treated rats with myocarditis) groups (n = 8). APA and APD at repolarizations of 20%, 50%, and 90% (APD20, APD50, and APD90) are indicated. B Superimposed traces depict APs in the RVAs of the control, experimental, and treatment groups (n = 8). APA, APD20, APD50, and APD90 are indicated. C Superimposed traces depict APs in the LVs of the control, experimental, and treatment groups (n = 8). APA, APD20, APD50, and APD90 are indicated. D Premature ventricular contractions and ventricular tachycardia in the RVOTs, RVAs, and LVs of the control (n = 6), experimental (n = 10), and treatment (n = 7) groups. AP, action potential; APA, action potential amplitude; APD, action potential duration; RVOT, right ventricle outflow tract; RVA, right ventricular apex; LV, left ventricle
Effects of myocarditis on RVOT electrical activity and intracellular ROS production
In the RVOTs of the experimental group, we observed several electrophysiological alterations. Specifically, the experimental group exhibited considerably larger INa-Late, larger reverse-mode NCX current, and smaller ICa-L than did the control group. However, MCC950-treated rats exhibited smaller INa-Late, lower NCX activity, and higher ICa-L than did the untreated experimental rats (Fig. 4). These findings suggest that MCC950 treatment can mitigate myocarditis-associated electrophysiological abnormalities in the RVOT of rats. Furthermore, the RVOTs of the experimental group had higher levels of intracellular and mitochondrial ROS than did the RVOTs of the control and treatment groups (Fig. 5 D,E). Therefore, MCC950 treatment mitigated the myocarditis-induced increase in oxidative stress within rat RVOTs. Similarly, the intracellular level of Na+ in the RVOTs of the experimental group was higher than that in the RVOTs of the control and treatment groups (Fig. 5F).

Estimates of INa-Late, ICa-L, and NCX current in rat RVOTs. A Tracings and I–V relationship of NCX current in the RVOT cardiomyocytes of the control (healthy rats; n = 10), experimental (rats with myocarditis; n = 10), and treatment (MCC950-treated rats with myocarditis; n = 9) groups. B Tracings and I–V relationship of ICa-L in the RVOT cardiomyocytes of the control (n = 14), experimental (n = 16), and treatment (n = 12) groups C Current tracings and average data of INa-Late in the RVOT cardiomyocytes of the control (n = 12), experimental (n = 12), and treatment (n = 8) groups. INa-Late, late Na+ current; ICa-L, L-type Ca2+ current; NCX, Na+–Ca2+ exchanger; RVOT, right ventricle outflow tract

ROS, intracellular Ca2+, SR Ca2+, Ca2+ leak, and cytosolic Na+ levels in rat RVOTs. A Tracings and average levels of [Ca2+]i transients and fractional SR Ca2+ release in the RVOT cardiomyocytes of the control (n = 30), experimental (rats with myocarditis; n = 28), and treatment (MCC950-treated rats with myocarditis; n = 30) groups. B Tracings and average SR Ca2+ levels in the RVOT cardiomyocytes of the control (n = 12), experimental (n = 12), and treatment (n = 12) groups. C Tracings and average Ca2+ leak levels in the RVOT cardiomyocytes of the control (n = 19), experimental (n = 14), and treatment (n = 15) groups. D Average levels of cytosolic ROS in the RVOT cardiomyocytes of the control (n = 30), experimental (n = 31), and treatment (n = 25) groups. E Average levels of mitochondrial ROS in the RVOT cardiomyocytes of the control (n = 33), experimental (n = 30), and treatment (n = 33) groups. F Average levels of cytosolic Na+ in the RVOT cardiomyocytes of the control (n = 30), experimental (n = 30), and treatment (n = 29) groups. ROS, reactive oxygen species; SR, sarcoplasmic reticulum; RVOT, right ventricle outflow tract
Effects of MCC950 on Ca2+ levels in rats with myocarditis
The RVOTs of the experimental group had smaller [Ca2+]i transients than did the RVOTs of the control and treatment groups. However, the control and treatment groups exhibited similar [Ca2+]i transients and SR Ca2+ levels (Fig. 5 A,B). To elucidate the underlying mechanisms, we measured the levels of Ca2+ in the SR and found that these levels were lower in the RVOTs of the experimental group than in the RVOTs of the control and treatment groups. The experimental group also exhibited a larger Ca2+ leak than did the control and treatment groups (Fig. 5C), suggesting that myocarditis reduces [Ca2+]i transients and SR Ca2+ levels by increasing Ca2+ leakage. These findings indicate that myocarditis results in reduced [Ca2+]i transients and SR Ca2+ levels in RVOT myocytes. Inhibition of NLRP3 signaling may mitigate the effects of myocarditis on Ca2+ handling and restore Ca2+ homeostasis.
Effects of MCC950 on Ca2+ regulatory proteins in rats with myocarditis
We investigated Ca2+ regulatory proteins that mediate the detrimental effects of myocarditis on Ca2+ homeostasis. The expression levels of NLRP3, NF-κB, IL-1B, RyR2, RyR2 phosphorylated at serine-2808 (pRyR2-S2808), and pCaMKII were higher but that of SERCA2a was lower in the RVOTs of the experimental group than in the RVOTs of the control group (Fig. 6). However, the expression levels of protein kinase A, phospholamban, Cav1.2, and phospholamban phosphorylated at T17 were similar in these two groups. After MCC950 treatment, we discovered upregulated expression of IL-1B, RyR2, pRyR2-S2808, and pCaMKII in the rat RVOTs (Fig. 6). Treatment of the experimental rats with anti-NLRP3 antibodies blocked the myocarditis-induced activation of Ca2+ signaling, implicating NLRP3 in the dysregulation of Ca2+ signaling in myocarditis. These findings indicate that myocarditis disrupts Ca2+ homeostasis in the RVOT by upregulating the expression of inflammatory proteins and Ca2+-handling proteins—such as NLRP3, RyR2, pRyR2-S2808, and pCaMKII—and by downregulating the expression of SERCA2a. Because these dysregulations were mitigated in the treatment group, we believe that MCC950 can be used to target NLRP3 to mitigate myocarditis-induced abnormalities in Ca2+ signaling.

Effects of the NLRP3/CaMKII axis on Ca2+ regulatory proteins in rat RVOTs. Representative Western blots and summary data for sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a, CaMKII phosphorylated at Thr286, RyR2, total phospholamban, RYR phosphorylated at Ser2808, phospholamban phosphorylated at Thr17, protein kinase A, Cav1.2, NLRP3, nuclear factor-κB, and interleukin-1β expression in the RVOT tissues of the control (healthy rats; n = 7), experimental (rats with myocarditis; n = 7), and treatment (MCC950-treated rats with myocarditis; n = 7) groups. Glyceraldehyde-3-phosphate dehydrogenase served as a loading control. CaMKII, Ca2 + /calmodulin-dependent protein kinase II; RYR, ryanodine receptor; RVOT, right ventricle outflow tract
Discussion
VT is a major cause of sudden cardiac death and mortality, both in the general population and patients with myocarditis [28, 29]. Nonsustained VT and frequent PVCs are associated with an increased risk of cardiovascular mortality [30]. Pathological data from the present study revealed that cardiac regions with higher levels of NLRP3 inflammasome activity are more susceptible to VA. This suggests a direct correlation between NLRP3 activity and arrhythmogenic events in rats with myocarditis. Our in vitro experiments indicated higher incidence of PVCs and VT in the experimental group than in the control group. In addition, we found that the RVOT is more susceptible than are other ventricular regions to myocarditis-induced arrhythmogenesis. This observation suggests that autoimmunity-induced myocarditis leads to arrhythmia in the RVOT, likely due to inflammation and electrolyte imbalance, particularly that involving abnormal Ca2+ handling. Notably, embryological studies have unraveled different origins of RVOT, RVA, and LV cardiomyocytes [31, 32]. The right ventricle, particularly the RVOT, may be more dependent than the LV on dynamic phosphorylation and dephosphorylation activities [33]. Our experimental group exhibited higher levels of NLRP3 expression and inflammation in the right ventricle than in the LV, and these higher levels potentially contributed to the increased susceptibility of the RVOT to myocarditis-associated arrhythmia. These findings elucidate the mechanisms underlying arrhythmogenesis in rats with myocarditis, highlighting the role of inflammation and NLRP3 activation, particularly in the RVOT. An improved understanding of these mechanisms may guide future therapeutic strategies for myocarditis-associated arrhythmia.
In patients with myocarditis, VA can occur at various stages of inflammation. During the acute phase, inflammation mediates the development of arrhythmogenic foci throughout the myocardium. Tissue edema increases the extracellular space, reducing the local myocardial conduction velocity in certain regions and thereby perhaps facilitating the formation of re-entry circuits [34, 35]. Abnormal cycling of Ca2+, downregulation of potassium channels, altered expression of gap junction proteins, and disruption of dystrophin increase the risk of VT by prolonging the duration of AP and creating arrhythmogenic conditions [34, 36]. In patients with late-onset VA after myocarditis, the regular and monomorphic type of VA is more consistent with stable scar-related re-entry circuits than are other types [8, 37]. The myocarditis-induced disruption of Ca2+ regulation in cardiac cells, which is exacerbated by caffeine exposure, can further predispose patients to arrhythmia [38]. In the present study, these effects were observed only in the experimental group, suggesting that the arrhythmic potential of caffeine is amplified by the combined effects of sympathetic activation and Ca2+ imbalance in rats with myocarditis. In myocarditis, NLRP3 expression increases as part of the immune response; we observed higher NLRP3 levels in the experimental and treatment groups than in the control group. MCC950 blocks the NLRP3 activator–induced release of IL‑1β [39,40,41] and inhibits the NLRP3 inflammasome by directly targeting the NATCH domain of NLRP3, thereby interfering with the function of the Walker B motif and preventing the conformational change and oligomerization of NLRP3 [42, 43]. Thus, MCC950 inhibits the activity, but not the expression, of the NLRP3 inflammasome [43]. Central to this process is NLRP3, which, upon activation, forms the inflammasome complex, leading to the release of proinflammatory cytokines such as IL-1β and IL-18. These cytokines play key roles in promoting the activation and proliferation of T cells, thus exacerbating the autoimmune response against cardiac myosin, resulting in myocardial inflammation and damage typical of myocarditis. In this study, we targeted NLRP3 signaling to prevent the activation of this inflammatory cascade and block the production of proinflammatory cytokines. Western blotting revealed elevated IL-1β levels in the experimental group, but the NLRP3 inhibitor MCC950 largely prevented the elevation of these levels. This finding underscores the potential of NLRP3 inhibition in managing myocarditis-induced inflammatory responses. We further found a more pronounced inflammatory response in the RVOT than in the LV, which is partly attributable to the higher contents of epicardial fat tissue in the RVOT [44, 45]. Adipose tissue in the heart mediates immune and inflammatory responses by secreting various cytokines and chemokines that amplify local inflammatory reactions [46, 47]. The RVOT typically has high content of adipose tissue, which is more prone to inflammation than are other types of tissue. The high content of adipose tissue in the RVOT may serve as a focal point for the infiltration of inflammatory cells and the production of cytokines in myocarditis. Thus, RVOT adipose tissue may be an immunologically active site and may have contributed to the regional heterogeneity observed in our study. In the experimental group, the triggered activity levels and VT incidence were significantly higher in the RVOTs than in did the RVAs and LVs. This finding suggests that in patients with acute myocarditis, VA is more likely to originate from the RVOT than from any other cardiac region. The incidence of VT was higher in the RVAs of the experimental group than in the RVAs of the control group; however, the difference was nonsignificant. This finding indicates the emergence of a persistent substrate for the formation of re-entry circuits, likely through replacement-type fibrosis and scar formation. However, myocarditis exerted a less pronounced effect on LV arrhythmogenesis, perhaps because of the large thickness of the LV myocardium. Compared with the RVOT, the LV has a thicker myocardium with a lower level of adipocyte infiltration, which buffers against the infiltration of inflammatory cells. These features of the LV contribute to its resistance to inflammation-induced arrhythmogenesis and enable it to withstand and recover from inflammation-induced stress and damage [48]. Rapamycin can induce a Ca2+ leak, thus increasing the risk of cardiac arrhythmia. This effect is attributable to rapamycin-mediated inhibition of the mechanistic target of rapamycin pathway, which is essential for maintaining the Ca2+ balance in cardiac myocytes. By disrupting the intracellular regulation of Ca2+, rapamycin triggers Ca2+ leakage and arrhythmogenesis [49, 50]. Our findings indicate variation in arrhythmogenicity across the cardiac regions of rats with myocarditis. The RVOT appears to be particularly susceptible to VA, whereas the RVA and LV exhibit varying degrees of susceptibility. Understanding the regional differences and the underlying pathophysiological mechanisms can facilitate the diagnosis and management of arrhythmia in patients with myocarditis.
COVID-19 can directly infect cardiomyocytes by attaching to angiotensin-converting enzyme 2 receptors, which are abundant in the heart, rendering the heart vulnerable to viral invasion [51]. Once inside the cells, the SARS-CoV-2 virus replicates and causes inflammation and damage to the cardiac muscle, potentially leading to myocarditis. In addition, the release of proinflammatory cytokines in response to COVID-19 results in widespread inflammation across organs, including the heart; this explains the association between COVID-19 and myocarditis [52]. COVID-19-associated myocarditis is a consequence of the virus’ presence and activity, mediated through inflammatory and immune responses, rather than of an autoimmune response, such as that observed in our animal model. Similar to COVID-19-associated myocarditis, autoimmunity-associated myocarditis results in upregulated activation of the NLRP3 and NF-κB pathways in addition to an increase in IL-1β level. This suggests common inflammatory mechanisms between COVID-19-associated and autoimmunity-associated myocarditis [51, 52]. Activation of the NLRP3 inflammasome drives the inflammatory response in both scenarios, leading to the secretion of cytokines. These findings indicate inflammation dynamics that are likely applicable to the cardiac implications of COVID-19. Patients with myocarditis commonly have prolonged QRS duration and QTc interval, which are attributable to an increase in the ventricular APD. This prolongation has been shown to render these patients susceptible to arrhythmogenesis [10, 53]. Prolonged QRS duration and QTc interval are associated with higher risks of fulminant disease and in-hospital mortality [53]. These findings corroborate ours: the QRS duration and QTc interval were longer in the experimental group than in the control and treatment groups. Furthermore, the APD was longer in the RVOTs of the experimental group than in the RVOTs of the control and treatment groups. A prolonged APD may increase the risk of VA in the RVOT, particularly in patients with long QT syndrome [54, 55]. Prolongation of APD further exacerbates the reductions in diastolic filling and stroke volume at high pulse rates [56]. A prolonged QT interval may increase the risk of VA by enhancing triggered activity, particularly through early depolarization [57]. Our findings revealed a more pronounced inflammatory response in the RVOT than in the RVA and LV, which may explain the observed differences in APD morphology. The RVOT may be more vulnerable to inflammatory stress because of its distinct histopathological characteristics, including its adipose tissue distribution. This finding is supported by that of a study indicating that inflammation can extend APD90 by modifying essential ion channels and Ca2+ handling [58]. The MCC950-mediated reduction in VA incidence in the RVOTs of the experimental group suggests that inhibiting NLRP3 with MCC950 reduces the risk of arrhythmia by suppressing arrhythmogenesis in the RVOT.
Our results indicated significantly larger SR Ca2+ leaks from the RVOTs of the experimental group than those of the control group. A larger Ca2+ leak may be associated with smaller [Ca2+]i transients and lower SR Ca2+ levels. The role of CaMKII as an intracellular Ca2+ sensor is particularly noteworthy in this context. CaMKII functions as a key regulator of immune and inflammatory responses, which mediate the alternations in Ca2+ handling [59, 60]. An increase in Ca2+ leakage is a key mechanism that contributes to the increased risk of arrhythmia in the RVOT, which emphasizes the role of Ca2+ dynamics in myocarditis-induced arrhythmogenesis [45]. In our study, the RVOT expression levels of RyR2, pRyR2 s2814, and pCaMKII were higher and that of SERCA2a was lower in the experimental group than in the control group. The RVOTs of the experimental group also had reduced SR Ca2+ levels and relatively large NCX current, which suggests a strong correlation between NCX current and Ca2+ homeostasis [61]. SERCA2a—which is modulated by phospholamban, sarcolipin, and CaMKII-mediated direct phosphorylation—facilitates the storage of Ca2+ within the SR [62]. Thus, the reuptake of cytosolic Ca2+ into the SR can be reduced by suppressing SERCA2a function. Hyperactive RyR–mediated leakage of Ca2+ from the SR may contribute to the incidence of VA [63]. Ca2+ overload can lead to inactivation of ICa-L in the RVOT, thereby inhibiting the influx of Ca2+ into cardiomyocytes [64]. In addition to the level of SERCA2a, that of Ca2+ in the SR is reduced due to the depletion of Ca2+ stores; eventually, a reduction is noted in [Ca2+]i transients because of the downregulation of the RYR2-mediated release of Ca2+ from the SR. In our study, the upregulated expression of pCaMKII in the RVOTs of the experimental group may have increased the leakage of Ca2+. Nonetheless, MCC95 mitigated the myocarditis-induced leakage of Ca2+; this finding suggests that activation of CaMKII is crucial for the effects of myocarditis on RVOT cardiomyocytes.
CaMKII substantially enhances INa-Late [65], thereby increasing intracellular Na+ loading and promoting arrhythmogenesis. Increased INa-Late has been implicated in the pathophysiology of acquired cardiac diseases, such as myocardial ischemia [66] and heart failure [67]. The present study revealed a significantly larger INa-Late in the RVOTs of the experimental group than in those of the control group. An increase in INa-Late may disrupt the balance between Na+ efflux and Ca2+ influx, resulting in intracellular Ca2+ overload [68]. However, in our study, MCC95-treated rats with myocarditis had smaller INa-Late than did their untreated counterparts. These findings suggest that MCC950 partially mitigates the risk of VA incidence in the RVOT by reducing INa-Late and thus highlight the roles of INa-Late and its MCC950-mediated modulation in the pathophysiology of myocarditis-associated arrhythmia. By targeting INa-Late and its downstream effects on intracellular Ca2+ handling, MCC950 may prevent arrhythmogenesis in the RVOTs of patients with myocarditis.
Activation of the NLRP3 inflammasome induces CaMKII signaling, which promotes cardiac remodeling [69]. Consistent with this finding, we observed significantly higher levels of NLRP3 inflammasome activation and CaMKII expression in the experimental group than in the control group. Inflammation-induced oxidative stress is a key mediator of myocarditis progression [70]. In our study, MCC950 downregulated the expression of ROS and CaMKII and the rate of arrhythmogenesis in the RVOTs of rats with myocarditis. These findings suggest that the activation of NLRP3-induced CaMKII signaling and the associated increase in ROS levels may contribute to the development of VA in rats with myocarditis. However, MCC950 effectively suppressed the activity of NLRP3, ROS, and CaMKII, thereby restoring NCX, INa-Late, and Ca2+ levels in the RVOTs of the experimental group (Fig. 7).

Potential mechanisms underlying the role of NLRP3 signaling in myocarditis. MCC950 treatment may reverse myocarditis-induced Na+–Ca2+ dysregulation by mitigating the alterations in levels of ROS, CMKII, RyR2, and ionic channels in cardiomyocytes. INa-Late: late Na+ current; NCX, Na+–Ca2+ exchanger; ROS, reactive oxygen species; RyR2, ryanodine receptor 2; RyR2-pS2808, RyR2 phosphorylated at serine 2808; SR, sarcoplasmic reticulum; SERCA, sarcoplasmic reticulum ATPase
Limitations
The present study has some limitations. First, this study primarily relied on a rat model of myosin peptide–induced myocarditis. Although this model provides valuable insights, myocarditis response may differ between rats and humans and vary depending on etiology. Therefore, our findings should be interpreted with caution when extrapolating to human myocarditis. Second, we administered MCC950 for a fixed duration (14 days). Further investigations into the optimal treatment duration and long-term effects of MCC950 are needed to enhance the clinical relevance of our findings. Third, this study primarily focused on NLRP3 signaling; however, myocarditis is a multifaceted condition influenced by various factors, such as viral infection, autoimmune response, and genetic predisposition. The contribution of these factors to RVOT arrhythmogenesis should not be overlooked. Fourth, our study mainly clarified the mechanisms underlying RVOT arrhythmogenesis. Future studies should investigate the clinical correlations of our findings with those observed in human patients with myocarditis. Finally, we studied arrhythmogenesis primarily in the RVOT, not the left ventricular outflow tract. This limitation necessitates further investigation.
Conclusion
Our findings suggest that RVOT cardiomyocytes are predisposed to myocarditis and associated arrhythmogenesis. Inhibition of NLRP3 may attenuate myocarditis-induced dysregulations in Ca2+ and Na+ levels by downregulating the expression of ROS and CaMKII, thereby normalizing key electrical and structural parameters. Therefore, in patients with myocarditis, NLRP3 signaling may be targeted to reduce the risk of VA.
Availability of data and materials
Not applicable.
Abbreviations
- AP:
-
Action potential
- APA:
-
Action potential amplitude
- APD:
-
Action potential duration
- Ca2+ :
-
Calcium
- CaMKII:
-
Ca2+/calmodulin-dependent protein kinase II
- ECG:
-
Electrocardiography
- I Na-Late :
-
Late sodium
- IL:
-
Interleukin
- LV:
-
Left ventricle
- NCX:
-
Na+–Ca2+ exchanger
- NLRP3:
-
Nucleotide oligomerization domain-like receptor family protein 3
- PVC:
-
Premature ventricular contraction
- RVA:
-
Right ventricular apex
- RVOT:
-
Right ventricular outflow tract
- ROS:
-
Reactive oxygen species
- RyR2:
-
Ryanodine receptor 2
- RyR2-pS2808:
-
RyR2 phosphorylated at serine 2808
- SR:
-
Sarcoplasmic reticulum
- SERCA:
-
Sarcoplasmic reticulum ATPase
- VA:
-
Ventricular arrhythmia
- VT:
-
Ventricular tachycardia
References
Gore I, Saphir O. Myocarditis; a classification of 1402 cases. Am Heart J. 1947;34:827–30.
Anzini M, Merlo M, Sabbadini G, Barbati G, Finocchiaro G, Pinamonti B, et al. Long-term evolution and prognostic stratification of biopsy-proven active myocarditis. Circulation. 2013;128:2384–94.
Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, et al. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2013;34:2636–48. 48a-48d.
Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, et al. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–84.
Wojnicz R, Nowalany-Kozielska E, Wojciechowska C, Glanowska G, Wilczewski P, Niklewski T, et al. Randomized, placebo-controlled study for immunosuppressive treatment of inflammatory dilated cardiomyopathy: two-year follow-up results. Circulation. 2001;104:39–45.
Mason JW, O’Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, et al. A clinical trial of immunosuppressive therapy for myocarditis. The myocarditis treatment trial investigators. N Engl J Med. 1995;333:269–75.
Lakkireddy D, Turagam MK, Yarlagadda B, Dar T, Hamblin M, Krause M, et al. Myocarditis causing premature ventricular contractions: Insights from the MAVERIC registry. Circ Arrhythm Electrophysiol. 2019;12:e007520.
Dello Russo A, Casella M, Pieroni M, Pelargonio G, Bartoletti S, Santangeli P, et al. Drug-refractory ventricular tachycardias after myocarditis: endocardial and epicardial radiofrequency catheter ablation. Circ Arrhythm Electrophysiol. 2012;5:492–8.
Tavazzi G, Pellegrini C, Maurelli M, Belliato M, Sciutti F, Bottazzi A, et al. Myocardial localization of coronavirus in COVID-19 cardiogenic shock. Eur J Heart Fail. 2020;22:911–5.
Etheridge SP, Asaki SY. COVID-19 infection and corrected QT interval prolongation-collateral damage from our newest enemy. JAMA Netw Open. 2021;4:e217192.
Lindner D, Fitzek A, Brauninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol. 2020;5:1281–5.
Lu YY, Chung FP, Chen YC, Tsai CF, Kao YH, Chao TF, et al. Distinctive electrophysiological characteristics of right ventricular out-flow tract cardiomyocytes. J Cell Mol Med. 2014;18:1540–8.
Lu YY, Cheng CC, Tsai CF, Lin YK, Lee TI, Chen YC, et al. Discrepant effects of heart failure on electrophysiological property in right ventricular outflow tract and left ventricular outflow tract cardiomyocytes. Clin Sci (Lond). 2017;131:1317–27.
Bao J, Sun T, Yue Y, **ong S. Macrophage NLRP3 inflammasome activated by CVB3 capsid proteins contributes to the development of viral myocarditis. Mol Immunol. 2019;114:41–8.
Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432–41.
Yao C, Veleva T, Scott L Jr, Cao S, Li L, Chen G, et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation. 2018;138:2227–42.
Myers JM, Fairweather D, Huber SA, Cunningham MW. Autoimmune myocarditis, valvulitis, and cardiomyopathy. Curr Protoc Immunol. 2013;15:15 4 1-51.
Chen KP, Hua KF, Tsai FT, Lin TY, Cheng CY, Yang DI, et al. A selective inhibitor of the NLRP3 inflammasome as a potential therapeutic approach for neuroprotection in a transgenic mouse model of Huntington’s disease. J Neuroinflammation. 2022;19:56.
Al-Qazazi R, Lima PDA, Prisco SZ, Potus F, Dasgupta A, Chen KH, et al. Macrophage-NLRP3 activation promotes right ventricle failure in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2022;206:608–24.
Lee TI, Chen YC, Lin YK, Chung CC, Lu YY, Kao YH, et al. Empagliflozin attenuates myocardial sodium and calcium dysregulation and reverses cardiac remodeling in streptozotocin-induced diabetic rats. Int J Mol Sci. 2019;20:1680.
Huang SY, Chen YC, Kao YH, Lu YY, Lin YK, Higa S, et al. Calcium dysregulation increases right ventricular outflow tract arrhythmogenesis in rabbit model of chronic kidney disease. J Cell Mol Med. 2021;25:11264–77.
Kao YH, Hsu JC, Chen YC, Lin YK, Lkhagva B, Chen SA, et al. ZFHX3 knockdown increases arrhythmogenesis and dysregulates calcium homeostasis in HL-1 atrial myocytes. Int J Cardiol. 2016;210:85–92.
Suenari K, Chen YC, Kao YH, Cheng CC, Lin YK, Chen YJ, et al. Discrepant electrophysiological characteristics and calcium homeostasis of left atrial anterior and posterior myocytes. Basic Res Cardiol. 2011;106:65–74.
Hove-Madsen L, Llach A, Bayes-Genis A, Roura S, Rodriguez Font E, Aris A, et al. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation. 2004;110:1358–63.
Huang SY, Chen YC, Kao YH, Hsieh MH, Lin YK, Chen SA, et al. Redox and activation of protein kinase a dysregulates calcium homeostasis in pulmonary vein cardiomyocytes of chronic kidney disease. J Am Heart Assoc. 2017;6:e005701.
Lu YY, Huang SY, Lin YK, Chen YC, Chen YA, Chen SA, et al. Epicardial adipose tissue modulates arrhythmogenesis in right ventricle outflow tract cardiomyocytes. Europace. 2021;23:970–7.
Kesavardhana S, Kanneganti TD. Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol. 2017;29:201–10.
Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–867.
Peretto G, Sala S, Rizzo S, Palmisano A, Esposito A, De Cobelli F, et al. Ventricular arrhythmias in myocarditis: characterization and relationships with myocardial inflammation. J Am Coll Cardiol. 2020;75:1046–57.
Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, et al. 2017 AHA/ACC/HRS guideline for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: executive summary: a report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines and the heart rhythm society. J Am Coll Cardiol. 2018;72:1677–749.
Moorman AF, Christoffels VM. Cardiac chamber formation: development, genes, and evolution. Physiol Rev. 2003;83:1223–67.
Chien KR, Domian IJ, Parker KK. Cardiogenesis and the complex biology of regenerative cardiovascular medicine. Science. 2008;322:1494–7.
DeGrande ST, Little SC, Nixon DJ, Wright P, Snyder J, Dun W, et al. Molecular mechanisms underlying cardiac protein phosphatase 2A regulation in heart. J Biol Chem. 2013;288:1032–46.
Veeraraghavan R, Salama ME, Poelzing S. Interstitial volume modulates the conduction velocity-gap junction relationship. Am J Physiol Heart Circ Physiol. 2012;302:H278–86.
Wakisaka Y, Niwano S, Niwano H, Saito J, Yoshida T, Hirasawa S, et al. Structural and electrical ventricular remodeling in rat acute myocarditis and subsequent heart failure. Cardiovasc Res. 2004;63:689–99.
Andreoletti L, Venteo L, Douche-Aourik F, Canas F, Lorin de la Grandmaison G, Jacques J, et al. Active Coxsackieviral B infection is associated with disruption of dystrophin in endomyocardial tissue of patients who died suddenly of acute myocardial infarction. J Am Coll Cardiol. 2007;50:2207–14.
Bhaskaran A, Tung R, Stevenson WG, Kumar S. Catheter ablation of VT in non-ischaemic cardiomyopathies: endocardial, epicardial and intramural approaches. Heart Lung Circ. 2019;28:84–101.
Kong H, Jones PP, Koop A, Zhang L, Duff HJ, Chen SR. Caffeine induces Ca2+ release by reducing the threshold for luminal Ca2+ activation of the ryanodine receptor. Biochem J. 2008;414:441–52.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21:248–55.
Zheng G, He F, Xu J, Hu J, Ge W, Ji X, et al. The selective NLRP3-inflammasome inhibitor MCC950 mitigates post-resuscitation myocardial dysfunction and improves survival in a rat model of cardiac arrest and resuscitation. Cardiovasc Drugs Ther. 2023;37:423–33.
Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol. 2019;15:556–9.
Tapia-Abellan A, Angosto-Bazarra D, Martinez-Banaclocha H, de Torre-Minguela C, Ceron-Carrasco JP, Perez-Sanchez H, et al. MCC950 closes the active conformation of NLRP3 to an inactive state. Nat Chem Biol. 2019;15:560–4.
Miles C, Westaby J, Ster IC, Asimaki A, Boardman P, Joshi A, et al. Morphometric characterization of collagen and fat in normal ventricular myocardium. Cardiovasc Pathol. 2020;48:107224.
Lu YY, Chen YC, Lin YK, Chen SA, Chen YJ. Electrical and structural insights into right ventricular outflow tract arrhythmogenesis. Int J Mol Sci. 2023;24:11795.
Anthony SR, Guarnieri AR, Gozdiff A, Helsley RN, Phillip Owens A, Tranter M. Mechanisms linking adipose tissue inflammation to cardiac hypertrophy and fibrosis. Clin Sci (Lond). 2019;133:2329–44.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96:939–49.
Zimmer A, Teixeira RB, Bonetto JHP, Bahr AC, Turck P, de Castro AL, et al. Role of inflammation, oxidative stress, and autonomic nervous system activation during the development of right and left cardiac remodeling in experimental pulmonary arterial hypertension. Mol Cell Biochem. 2020;464:93–109.
Ivarsson N, Mattsson CM, Cheng AJ, Bruton JD, Ekblom B, Lanner JT, et al. SR Ca(2+) leak in skeletal muscle fibers acts as an intracellular signal to increase fatigue resistance. J Gen Physiol. 2019;151:567–77.
Decuypere JP, Kindt D, Luyten T, Welkenhuyzen K, Missiaen L, De Smedt H, et al. mTOR-controlled autophagy requires intracellular Ca(2+) signaling. PLoS ONE. 2013;8:e61020.
Pannucci P, Jefferson SR, Hampshire J, Cooper SL, Hill SJ, Woolard J. COVID-19-induced myocarditis: pathophysiological roles of ACE2 and toll-like receptors. Int J Mol Sci. 2023;24:5374.
Fairweather D, Beetler DJ, Di Florio DN, Musigk N, Heidecker B, Cooper LT Jr. COVID-19, myocarditis and pericarditis. Circ Res. 2023;132:1302–19.
Hung Y, Lin WH, Lin CS, Cheng SM, Tsai TN, Yang SP, et al. The prognostic role of QTc interval in acute myocarditis. Acta Cardiol Sin. 2016;32:223–30.
Haissaguerre M, Extramiana F, Hocini M, Cauchemez B, Jais P, Cabrera JA, et al. Map** and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation. 2003;108:925–8.
Bonatti V, Rolli A, Botti G. Recording of monophasic action potentials of the right ventricle in long QT syndromes complicated by severe ventricular arrhythmias. Eur Heart J. 1983;4:168–79.
Lee TI, Chen YC, Kao YH, Hsiao FC, Lin YK, Chen YJ. Rosiglitazone induces arrhythmogenesis in diabetic hypertensive rats with calcium handling alteration. Int J Cardiol. 2013;165:299–307.
Yan GX, Wu Y, Liu T, Wang J, Marinchak RA, Kowey PR. Phase 2 early afterdepolarization as a trigger of polymorphic ventricular tachycardia in acquired long-QT syndrome : direct evidence from intracellular recordings in the intact left ventricular wall. Circulation. 2001;103:2851–6.
Bi X, Zhang S, Jiang H, Ma W, Li Y, Lu W, et al. Mechanistic insights into inflammation-induced arrhythmias: a simulation study. Front Physiol. 2022;13:843292.
Nghiem P, Ollick T, Gardner P, Schulman H. Interleukin-2 transcriptional block by multifunctional Ca2+/calmodulin kinase. Nature. 1994;371:347–50.
Lin MY, Zal T, Ch’en IL, Gascoigne NR, Hedrick SM. A pivotal role for the multifunctional calcium/calmodulin-dependent protein kinase II in T cells: from activation to unresponsiveness. J Immunol. 2005;174:5583–92.
Hobai IA, O’Rourke B. Enhanced Ca(2+)-activated Na(+)-Ca(2+) exchange activity in canine pacing-induced heart failure. Circ Res. 2000;87:690–8.
Frank KF, Bolck B, Erdmann E, Schwinger RH. Sarcoplasmic reticulum Ca2+-ATPase modulates cardiac contraction and relaxation. Cardiovasc Res. 2003;57:20–7.
Fauconnier J, Meli AC, Thireau J, Roberge S, Shan J, Sassi Y, et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc Natl Acad Sci U S A. 2011;108:13258–63.
Kubalova Z. Inactivation of L-type calcium channels in cardiomyocytes. Experimental and theoretical approaches. Gen Physiol Biophys. 2003;22:441–54.
Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. 2019;127:246–59.
Maier LS, Sossalla S. The late Na current as a therapeutic target: where are we? J Mol Cell Cardiol. 2013;61:44–50.
Pourrier M, Williams S, McAfee D, Belardinelli L, Fedida D. CrossTalk proposal: The late sodium current is an important player in the development of diastolic heart failure (heart failure with a preserved ejection fraction). J Physiol. 2014;592:411–4.
Bers DM, Bassani JW, Bassani RA. Na-Ca exchange and Ca fluxes during contraction and relaxation in mammalian ventricular muscle. Ann N Y Acad Sci. 1996;779:430–42.
Suetomi T, Willeford A, Brand CS, Cho Y, Ross RS, Miyamoto S, et al. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca(2+)/Calmodulin-dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation. 2018;138:2530–44.
Wang Y, Gao B, **ong S. Involvement of NLRP3 inflammasome in CVB3-induced viral myocarditis. Am J Physiol Heart Circ Physiol. 2014;307:H1438–47.
Acknowledgements
Not applicable.
Funding
This work was supported by the Ministry of Science and Technology (grant numbers: MOST109-2314-B-038–124-MY3, MOST110-2314-B-038–107-MY3, MOST110-2314-B-016–037-MY3, MOST110-2314-B-075–063-MY3, MOST111-2314-B-038–158, and NSTC112-2314-B-038–124-MY3), Taipei Medical University–Wan Fang Hospital (grant numbers: 109-wf-eva-04, 109-wf-eva-18, 109-wf-swf-09, and 112-wf-phd-04), and the Ministry of National Defense-Medical Affairs Bureau, Taiwan (grant number: MND-MAB-D-113144).
Author information
Authors and Affiliations
Contributions
Conceptualization, CGC and YJC; data curation, FJL; formal analysis, YYL and YKL; investigation, TYC; methodology, CGC, YJC, and YCC; project administration, YJC; visualization, YKL; writing—original draft, CGC; writing—review & editing, YJC and SAC. All authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Institutional Animal Care and Use Committee of Taipei Medical University (animal use permission number: LAC-2020–0416).
Consent for publication
Not applicable.
Competing interests
All authors declare no competing interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chin, CG., Chen, YC., Lin, FJ. et al. Targeting NLRP3 signaling reduces myocarditis-induced arrhythmogenesis and cardiac remodeling. J Biomed Sci 31, 42 (2024). https://doi.org/10.1186/s12929-024-01032-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-024-01032-7