
Abstract
Background
The GNAS gene on chromosome 20q13.3, encodes the alpha-subunit of the stimulatory G protein, which is expressed in most tissues and regulated through reciprocal genomic imprinting. Disorders of GNAS inactivation produce several different clinical phenotypes including pseudohypoparathyroidism (PHP), pseudopseudohypoparathyroidism (PPHP), progressive osseous heteroplasia (POH), and osteoma cutis (OC). The clinical and biochemical characteristics overlap of PHP subtypes and other related disorders presents challenges for differential diagnosis.
Methods
We enrolled a total of 11 Chinese children with PHP in our study and analyzed their clinical characteristics, laboratory results, and genetic mutations.
Results
Among these 11 patients, nine of them (9/11) presented with resistance to parathyroid hormone (PTH); and nine (9/11) presented with an Albright′s hereditary osteodystrophy (AHO) phenotype. GNAS abnormalities were detected in all 11 patients, including nine cases with GNAS gene variations and two cases with GNAS methylation defects. These GNAS variations included an intronic mutation (c.212 + 3_212 + 6delAAGT), three missense mutations (c.314C > T, c.308 T > C, c.1123G > T), two deletion mutations (c.565_568delGACT*2, c.74delA), and two splicing mutations (c.721 + 1G > A, c.432 + 1G > A). Three of these mutations, namely, c.314C > T, c.1123G > T, and c.721 + 1G > A, were found to be novel. This data was then used to assign a GNAS subtype to each of these patients with six cases diagnosed as PHP1a, two cases as PHP1b, one as PPHP, and two as POH.
Conclusions
Evaluating patients with PTH resistance and AHO phenotype improved the genetic diagnosis of GNAS mutations significantly. In addition, our results suggest that when GNAS gene sequencing is negative, GNAS methylation study should be performed. Early genetic detection is required for the differential diagnosis of GNAS disorders and is critical to the clinician’s ability to distinguish between heterotopic ossification in the POH and AHO phenotype.
Similar content being viewed by others


Background
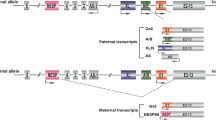
The GNAS gene encodes the alpha-subunit of the stimulatory G protein (Gsα) as well as four other transcripts. This gene exhibits parent-specific methylation in its promoters and some transcripts. GNAS also experiences a tissue-specific reduction in the expression of the paternal GNAS gene in some tissues including the proximal renal tubules, pituitary gland, thyroid, and gonads [1]. Several hormones are regulated via the signal transduction pathway downstream of Gsα and cyclic adenosine monophosphate (cAMP) [2]. Inactivating mutations in exons 1–13 of the GNAS gene can result in the expression of a defective Gsα protein, which can cause Albright’s hereditary osteodystrophy (AHO) that presents as tissue resistance to several hormones and distinct skeletal and developmental defects [3].
Pseudohypoparathyroidism (PHP) consists of a heterogeneous group of disorders that exhibit one common feature, namely, resistance to the actions of the parathyroid hormone (PTH). These disorders are roughly classified into PHP types 1 and 2. PHP type 1 is then further classified into three subtypes, namely, 1a, 1b, and 1c based on their clinical presentation, GNAS variant, and various laboratory findings, including their response to exogenous PTH infusion and Gsα activity. When heterozygous mutations are located in the maternal allele of GNAS exons 1–13, patients exhibited multiple hormone resistance, which act via the Gs-coupled receptors such as parathyroid hormone (PTH), thyroid-stimulating hormone (TSH), gonadotropins, growth hormone-releasing hormone (GHRH), in addition to the more traditional AHO features. This disorder is defined as PHP1a [1]. In contrast, PHP1b is caused by epigenetic abnormalities within differentially methylated regions (DMRs) of GNAS which may affect several transcripts, including Gsα, XLas, NESP55, 1A, and antisense (AS) [4]. PHP1b is characterized by resistance to PTH and other hormones. However, these patients do not exhibit AHO phenotypes [1]. PHP1c is caused by receptor coupling defects in the same Gsα and cAMP signal transduction pathway as PHP1a and presents with the same clinical features and laboratory findings [5], but most patients in this group do not present with any of the known pathogenic mutations of the Gsα encoding exons of the GNAS gene. Previous studies provided evidence that an overlap exists between the clinical features of different PHP subtypes [6].
Patients more frequently exhibit the AHO phenotype, without hormone resistance, when the same GNAS mutations are found on the paternal allele. These conditions are often referred to as pseudopseudohypoparathyroidism (PPHP) [7]. The AHO phenotype includes several physical characteristics, including brachydactyly, stocky build, short stature, obesity, round face, ectopic ossifications, and intellectual disability [1]. These features can also occur in both PHP and PPHP with variable intensity. Gsα haploinsufficiency is thought to play a primary role in the development of the AHO phenotype [2].
Progressive osseous heteroplasia (POH) is another ultra-rare genetic disease caused by heterozygous inactivating mutations in GNAS exons 1–13 and is characterized by cutaneous ossifications that proceed into the subcutaneous and deep connective tissues, including muscles, tendons, and ligaments [8]. POH is not however associated with either hormone resistance or the AHO phenotype.
This study evaluated the clinical and genetic characteristics of 11 Chinese children with clinical presentations consistent with GNAS spectrum disorders. Our evaluation was largely consistent with previous reports, and of the 11 mutations identified in this study, three were shown to be novel. Because of the overlap between the clinical and biochemical features of different PHP subtypes, accurate diagnosis requires a careful molecular and epigenetic analysis of GNAS. Furthermore, two patients were affected by PHP1a and PPHP and were treated with growth hormone (GH) therapy, which provide new data for GH therapy in PHP patients without GH deficiency.
Methods
Patients and Clinal data collection
A total of 11 cases of PHP were identified and enrolled in our study at the Shanghai Children's Medical Center, Shanghai Jiaotong University School of Medicine, between July 2017 and May 2021. The documented medical history of each patient including the status of their birth, growth, development, past illness, phenotypes, and family history were preserved for use in this study. Both routine and specific biochemical tests, including electrolyte and 25-hydroxy vitamin D, were performed on each patient. Their PTH, thyroid hormones, insulin-like growth factor-1(IGF-1), and adrenocorticotropic hormone (ACTH) indices were also evaluated. Each patient was also subject to x-ray examination and Cranial MRI or CT, with the exception of patient 2 and patient 5.
Ethical approval for this study was obtained from the ethics committee of the Shanghai Children's Medical Center and informed consent for genetic analysis and the collection of clinical data were obtained from the participant’s healthcare proxies.
Genetic analysis
Genomic DNA was extracted from the peripheral blood samples collected from each of these 11 patients and their parents using a QIAamp Blood DNA Mini Kit® (Qiagen GmbH, Hilden, Germany). Targeted next-generation sequencing and data analysis were then performed as described in our previous study [9]. All suspected variants were confirmed by Sanger sequencing and validated by parental testing. Manual classification of these variants was then completed using the method recommended by the American College of Medical Genetics and Genomics (ACMG) [10]. Potential pathogenicity of novel missense variants was determined using three in silico prediction methods, including Mutation Taster (http://www.mutationtaster.org), Sorting Tolerant from Intolerant (SIFT; http://sift.jcvi.org/), and Polymorphism Phenoty** v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/).
Certain samples that were negative for GNAS mutation when evaluated by Next generation sequencing (NGS) were then subjected to GNAS methylation detection. This methylation analysis was performed using methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) and the ME031A kit (MRCHolland, Amsterdam, The Netherlands) as per the manufacturer’s instructions. MS-MLPA data was then analyzed using Coffalyser software (MRC‐Holland, the Netherlands).
Results
Clinical description and laboratory results for each of the 11 patients in this study
We analyzed the phenotype and genotype of 11 Chinese children with PHP from 11 different families, with no consanguineous marriages. There were six males and five females in our cohort with a male to female ratio of 1.2:1. The age at presentation ranged from neonates to 13 years old, and the age at diagnosis ranged from 6 months to 14 years (the average being 6.5 ± 5.2 years). Among the 11 patients, three cases were referred to our clinic due to heterotopic ossification (P1, P10, P11), four cases for recurrent seizures (P3, P4, P7, P8), and five cases for reduced stature (P2, P4, P5, P6, P9). P6 received growth hormone treatment due to a reduced growth rate, and P9 was previously diagnosed as having idiopathic short stature (ISS) in a local hospital and was also given recombinant human growth hormone (rhGH) therapy. P3 presented with a familial history of AHO-like features. However, patient P3’s mother had no hormone resistance, and was diagnosed as PPHP. These patients’ clinical features are summarized in Table1 and Fig. 1.

The clinical characteristics of patients with pseudohypoparathyroidism. a High density shadows in the subcutaneous soft tissue of both the wrist and right elbow were observed in patient 1.b Brachydactyly of the hand and feet digits was presented in patient 2. Shortening of the metacarpals and metatarsals was revealed by X-ray, involving F2-5 on the left, F3-5 on right, and T3-4 on both sides. c Brachydactyly of the hands and feet was noted in patient 3. Radiographs of patient 3 revealed shortening of the metacarpals and metatarsals, particularly F1-5 and T4 on both sides. Patient 3 also presented with subcutaneous calcifications across their back, and cranial CT revealed multiple bilateral calcifications involving the cerebral hemisphere. d Patient 6 presented with a rounded face, and her head MRI revealed basal ganglia calcification. e Patient 9 experienced deformity of thorax, and her chest radiograph revealed scoliosis. f Patient 10 suffered from subcutaneous calcifications in the left thigh with develo** nodes in the left wrist, both shanks, the back, the abdomen, the auricula, and the inframandibular region. Radiograph revealed high density shadows in both lower limbs
A total of 54.5% (six cases) of our cohort were diagnosed with PHP1a, 18.2% (2 cases) with PHP1b, 9% (one case) with PPHP, and 18.2% (two cases) with POH. The clinical and laboratory data for each of the 11 cases are summarized in Table 1. All six cases (P1–P6) of PHP1a exhibited features of AHO (shown in Fig. 1). Laboratory tests revealed PTH resistance in all six patients with three cases (P3, P4, P5) presenting with hypocalcemia, five (P1, P3, P4, P5, P6) with TSH resistance, and two cases (P1, P4) with mildly elevated plasma ACTH. Regular X-rays of P1 revealed multiple ossifications in her left upper limb (Fig. 1a), and cranial CT identified multiple bilateral calcifications of the cerebral hemisphere in P3 and P6 (Fig. 1c, 1d).
The two PHP1b cases (P7, P8) presented with recurrent seizures due to hypocalcemia; neither exhibited any symptoms of AHO, presenting with only PTH resistance. Cranial CT of P7 revealed multiple high-density changes, while P9, who was diagnosed with PPHP, demonstrated a mild AHO phenotype, including short stature, brachydactyly and deformity of thorax (Fig. 1e). Because of short stature, P9 was subject to GH stimulation testing which revealed that their GH peak was normal (10.9 µg/µL, 4.7 µg/µL). Moreover, GH treatment has no evident effect. The two POH patients (P10, P11) presented with heterotopic ossification, involving both skin and muscle (Fig. 1f) and demonstrated no hormone resistance. A summary of these laboratory and imaging results is recorded in Table 2.
Identification of GNAS gene abnormalities
Of the 11 PHP patients in this study, nine presented with variations in the GNAS gene and two with GNAS methylation defects (Table 2). The GNAS gene variations included an intronic mutation (c.212 + 3_212 + 6 delAAGT), three missense mutations (c.314 C > T, c.308 T > C, and c.1123G > T), two deletion mutations (c.565_568delGACT and c.74delA), and two splicing mutations (c.721 + 1G > A and c.432 + 1G > A). Three of these mutations were found to be novel, namely, c.314C > T, c.1123G > T, and c.721 + 1G > A. No mutation of exons 1–13 of GNAS were identified in P7 and P8. However, we did detect a loss of methylation in the XLas and NESPAS regions of this gene in P7, and a loss of methylation in the XLas and AS region of this gene in P8.
Discussion
All of the patients recruited to this study presented with clinical features related to GNAS inactivation. We classified these patients into different disease types based on their clinical and molecular evaluations and performed genotype–phenotype correlation analysis. This revealed that patients with PTH resistance, such as P3, may present with hyperphosphatemia, hypocalcemia, and muscle weakness. Despite the presence of PTH resistance in patients 1 and 2, some cases might have elevated PTH levels with normal serum calcium and phosphate levels [11]. There was no resistance to the effects of PTH in the bone or ascending tubules of these patients that could explain this outcome [12]. Other subtypes of PHP could be distinguished from PHP1a based on these features. Furthermore, subclinical hypothyroidism was a hallmark of PHP1a [13]. Both patients, P1 and P3, experienced elevated thyroid stimulating hormone (TSH) levels with normal free thyroxine 4 (FT4) levels, suggesting that both of these patients were suffering from subclinical hypothyroidism. Patients P4, P5, and P6 presented with increased TSH and decreased FT4, which indicated TSH resistance, and all three received L-Thyroxine therapy.
The AHO phenotype, which is characterized by both skeletal and developmental defects, is a group of physical characteristics with variable expressivities. A clinical review showed that there were two different periods characterized by: 1) round face, rapid weight gain, subclinical hypothyroidism, and subcutaneous calcifications in toddlers, and 2) moderate intellectual disability, brachydactyly, afebrile seizures (hypocalcemia), short stature, and TSH resistance in older children [12]. The presentation of our patients were consistent with the above clinical studies. In addition, brachydactyly and heterotopic ossifications have been thought to be the most distinctive feature of the AHO phenotype [13]. Moreover, we observed strabismus in patient 3 and 9; this symptom has been reported in other studies and may represent an additional AHO-related feature [14].
PHP1b is characterized by PTH resistance, hypocalcemia, and hyperphosphatemia, without the addition of AHO-like symptoms. PHP1b is caused by epigenetic changes of the DMR within GNAS. Approximately 80% of PHP1b patients present with a sporadic mutation, and only 20% are autosomal dominant (AD) maternally inherited [15]. Both patients P7 and P8 experienced some form of hypocalcemia at 14 and 2 years old, respectively. These patients had been evaluated at the local hospital using NGS, but the results of these evaluations were negative. Given this, we went on to perform GNAS methylation detection, and the diagnosis of PHP1b was finally confirmed. This data suggests that patients who present with both PTH resistance and hypocalcemia and without AHO phenotypes, should be routinely evaluated by GNAS methylation detection as the first line of testing.
With the exception of those with PHP1b, a large proportion of PHP patients experience short stature into adulthood. This is likely a result of their GHRH resistance, with 50% – 80% of patients with PHP1a presenting with some degree of GH deficiency [16, 17]. The efficacy of rhGH treatment in patients with PHP1a was first studied in 2010, with the evaluation of changes in height velocity in eight prepubertal patients with PHP1A who had GH deficiency following rhGH therapy [18]. P6, who had PHP1a, experienced an increase in their growth rate in response to GH treatment, but P9, who had PPHP and was initially diagnosed with ISS, demonstrated no response to their rhGH therapy. The first international Consensus Statement for PHP recommends that patients with GH deficiencies should be treated with rhGH [19], but its use in patients without GH deficiency needs further evaluation.
Heterotopic ossification in POH can be distinguished from AHO by the presence of progressive ossification moving from subcutaneous tissues into deep connective tissues, as shown in patients P10 and P11. Heterotopic ossification can result in the ankylosis of affected joints and lead to severe disability; follow-up is needed to monitor the progression of this ossification. POH should also be distinguished from other genetic conditions causing heterotopic ossification such as fibro dysplasia ossificans progressive (FOP). These patients did not present with the congenital malformations of the large toes and pre-osseous tumor-like inflammation or “flare-ups” associated with FOP, which aided in their differential diagnosis [20].
The first mutation in GNAS was reported in 1990 [21] with numerous other mutations described since then. These include missense mutations, splicing substitutions, small/cross deletions or insertions, regulatory substitutions, small indels, and complex rearrangements. A small 4-bp deletion at codons 189–190 in exon 7 (c.565_568delGACT) of GNAS has been confirmed to be a mutational hot spot [22], and this hot spot was present in two of our patients (P3 and P5). We also identified a heterozygous 4-bp deletion mutation (c.212 + 3_212 + 6delAAGT) in P1, which is predicted to disrupt the highly conserved 5' splice site sequence in intron 2 and activate a cryptic splice site located 28 bp downstream of intron 2 [23]. P2 had a novel missense variant, c.314C > T(p.Thr105Ile), in exon 5. Exon 5 of the GNAS gene encodes the adenylate cyclase activation domain of Gsα, in which amino acids 70 to 140 have been reported to be important for functionality in activating adenylyl cyclase [24]. P4 had a novel nucleotide exchange c.1123G > T in exon 12, leading to a Valine to Leucine substitution (p.Val375Leu). Furthermore, multi-bioinformatics in silico software predicted that these variants have deleterious effects, and both variants were classified as likely pathogenic using the ACMG guidelines. Patient 10 had a novel splicing variant (c.721 + 1 G > A) in exon 9, which occurs within the + 1 splice site (PSV1) and was de novo (PS2), indicating that this variant is also pathogenic (PSV1 + PS2 + PM2 + PP4). P6 presented with a c.432 + 1G > A mutation; however, the same splicing mutation was reported in a boy with recurrent medulloblastoma [25], indicating that our patient had a risk of develo** tumors, suggesting that she will need careful monitoring and that her rhGH treatment should be more conservative.
Our study was a retrospective study from a single center, and the sample size was small. Large sample studies are needed to broaden the range of our results. Moreover, the effect of rhGH therapy in PPHP or other types of PHP, should be further investigated.
Conclusions
We completed a phenotypic and molecular assessment of 11 patients with PHP, which can be difficult to diagnose as its clinical phenotype is highly variable, and Gsα activity is not routinely assessed or available. Therefore, NGS and methylation detection of GNAS serve as reliable methods of confirming PHP diagnosis. We demonstrate that patients who present with PTH resistance and no AHO phenotype should be evaluated for changes in GNAS methylation to aid in differential diagnosis. In addition, our data supports the conclusion that it is necessary for clinicians to distinguish between the heterotopic ossification associated with POH and the AHO phenotype. This study not only expands our understanding of the phenotypic spectrum of GNAS mutations, but also deepens our understanding of the clinical phenotype associated with alterations in the GNAS gene.
Availability of data and materials
All the data during this study are included in this published article.
Abbreviations
- PHP:
-
Pseudohypoparathyroidism
- PPHP:
-
Pseudopseudohypoparathyroidism
- POH:
-
Progressive osseous heteroplasia
- OC:
-
Osteoma cutis
- PTH:
-
Parathyroid hormone
- AHO:
-
Albright′s hereditary osteodystrophy
- Gsα:
-
Alpha-subunit of the stimulatory G protein
- cAMP:
-
Cyclic adenosine monophosphate
- TSH:
-
Thyroid-stimulating hormone
- GHRH:
-
Growth hormone-releasing hormone
- DMRs:
-
Differentially methylated regions
- AS:
-
Antisense
- GH:
-
Growth hormone
- IGF-1:
-
Insulin-like growth factor-1
- ACTH:
-
Adrenocorticotropic hormone
- NGS:
-
Next generation sequencing
- MS-MLPA:
-
Methylation-specific multiplex ligation-dependent probe amplification
- ISS:
-
Idiopathic short stature
- rhGH:
-
Recombinant human growth hormone
- TSH:
-
Thyroid stimulating hormone
- FT4:
-
Free thyroxine 4
- AD:
-
Autosomal dominant
- FOP:
-
Fibro dysplasia ossificans progressive
References
Tafaj O, Jüppner H. Pseudohypoparathyroidism: one gene, several syndromes. J Endocrinol Invest. 2017;40:347–56.
Turan S, Bastepe M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. J Horm Res Paediatr. 2013;80:229–41.
Demiral M, Bozdağ Ö, Karaer K. A novel mutation in a case of pseudohypoparathyroidism type Ia Turk J Pediatr. 2016;58:101–5.
Turan S, Bastepe M. The GNAS complex locus and human diseases associated with loss-of-function mutations or epimutations within this imprinted gene. Horm Res Paediatr. 2013;80:229–41.
Elli FM, de Sanctis L, Peverelli E, Bordogna P, Pivetta B, Miolo G, Beck-Peccoz P, Spada A, Mantovani G. Autosomal dominant pseudohypoparathyroidism type Ib: a novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J Clin Endocrinol Metab. 2014;99:724–8.
Fernández-Rebollo E, Lecumberri B, Gaztambide S, Martinez-Indart L, de Nanclares Perez G, Castaño L, Spanish PHP Group. Endocrine profile and phenotype-(epi)genotype correlation in Spanish patients with pseudohypoparathyroidism. J Clin Endocrinol Metab. 2013;98(5):E996-1006.
Benvenuto P, Attarian A. Pseudopseudohypoparathyroidism: a diagnostic consideration in a patient with Brachydactyly. J Pediatr. 2018;196:321.
Grigelioniene G, Nevalainen PI, Reyes M, Thiele S, Tafaj O, Molinaro A, Takatani R, Ala-Houhala M, Nilsson D, Eisfeldt J, Lindstrand A, Kottler ML, Mäkitie O, Jüppner H. A large inversion involving GNAS exon A/B and all exons encoding Gsα is associated with autosomal dominant pseudohypoparathyroidism type Ib (PHP1b). J Bone Miner Res. 2017;32:776–83.
Li N, Chang G, Yufei Xu, Ding Yu, Li G, Tingting Yu, Qing Y, Li J, Shen Y, Wang J, Wang X. Clinical and molecular characterization of patients with Fructose 1,6-Bisphosphatase Deficiency. Int J Mol Sci. 2017;18:857.
Richards S, Aziz N, Bale S. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Levine MA. An update on the clinical and molecular characteristics of pseudohypoparathyroidism. Curr Opin Endocrinol Diabetes Obes. 2012;19:443–51.
Kayemba-Kay’s S, Tripon C, Heron A, Hindmarsh P. Pseudohypoparathyroidism type 1a-subclinical hypothyroidism and rapid weight gain as early clinical signs: a clinical review of 10 cases. J Clin Res Pediatr Endocrinol. 2016;8:432–8.
Mantovani G. Clinical review: pseudohypoparathyroidism: diagnosis and treatment. J Clin Endocrinol Metab. 2011;96:3020–30.
Sano S, Nakamura A, Matsubara K, Nagasaki K, Fukami M, Kagami M, Ogata T. (Epi)genotype-Phenotype Analysis in 69 Japanese Patients with Pseudohypoparathyroidism Type I. J Endocr Soc. 2018;2:9–23.
Elli FM, Linglart A, Garin I, Sanctis L, Bordogna P, Grybek V, Pereda A, Giachero F, Verrua E, Hanna P, Mantovani G, Perez de Nanclares G. The prevalence of GNAS deficiency-related diseases in a large cohort of patients characterized by the EuroPHP network. J Clin Endocrinol Metab. 2016;101:3657–68.
Germain-Lee EL, Groman J, Crane JL, de Beur Jan SM, Levine MA. Growth hormone deficiency in pseudohypoparathyroidism type 1a: another manifestation of multihormone resistance. J Clin Endocrinol Metab. 2003;88:4059–69.
Mantovani G, Maghnie M, Weber G, De Menis E, Brunelli V, Cappa M, Loli P, Beck-Peccoz P, Spada A. Growth hormone-releasing hormone resistance in pseudohypoparathyroidism type ia: new evidence for imprinting of the Gs alpha gene. J Clin Endocrinol Metab. 2003;88:4070–4.
Mantovani G, Ferrante E, Giavoli C, Linglart A, Cappa M, Cisternino M, Maghnie M, Ghizzoni L, De Sanctis L, Lania AG, Beck-Peccoz P, Spada A. Recombinant human GH replacement therapy in children with pseudohypoparathyroidism type Ia: first study on the effect on growth. J Clin Endocrinol Metab. 2010;95:5011–7.
Mantovani G, Bastepe M, Monk D, De Sanctis L, Thiele S, Usardi A, Ahmed SF, Bufo R, Choplin T, De Filippo G, Devernois G, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. 2018;14(8):476–500.
Kaplan FS, Shore EM. Progressive osseous heteroplasia. J Bone Miner Res. 2000;15:2084–94.
Patten JL, Johns DR, Valle D, Eil C, Gruppuso PA, Steele G, Smallwood PM, Levine MA. Mutation in the gene encoding the stimulatory G protein of adenylate cyclase in Albright’s hereditary osteodystrophy. N Engl J Med. 1990;322:1412–9.
Hainline BE, Brener JL, Wilson KA, Wilson LC, Oude-Luttikhuis ME, Trembath RC, Weinstein LS. A deletion hot-spot in exon 7 of the Gsa gene (GNAS1) in patients with Albright hereditary osteodystrophy. Hum Mol Genet. 1995;4:2001–2.
De Sanctis L, Romagnolo D, Olivero M, Buzi F, Maghnie M, Scirè G, Crino A, Baroncelli GI, Salerno M, Di Maio S, Cappa M, Grosso S, Rigon F, Lala R, De Sanctis C, Dianzani I. Molecular analysis of the GNAS1 gene for the correct diagnosis of Albright hereditary osteodystrophy and pseudohypoparathyroidism. Pediatr Res. 2003;53:749–55.
Antonelli M, Birnbaumer L, Allende JE, Olate J. Human-Xenopus chimeras of Gsα reveal a new region important for its activation of adenylyl cyclase. FEBS Lett. 1994;340:249–54.
Tokita MJ, Nahas S, Briggs B, Malicki DM, Mesirov JP, Reyes IA, Farnaes L, Levy ML, Kingsmore SF, Dimmock D, Crawford JR, Wechsler-Reya RJ. Biallelic loss of GNAS in a patient with pediatric medulloblastoma. Cold Spring Harb Mol Case Stud. 2019;5:a004572.
Acknowledgements
We acknowledge the patients and their parents for participating in this study.
Funding
This work was supported by National Science Foundation for Young Scientists of China (Grant No.81900722) and the Project of Shanghai Municipal Science and Technology Commission (20MC1920400) support this study.
Author information
Authors and Affiliations
Contributions
The first two authors were equally responsible for the work described in this paper. G.C., Q.L. should be considered joint first author. X.W., J.W. contributed to the conception and design, revising the manuscript. J.L., Y.D., X.H., Y.S., contributed to provision of study materials or patients. N.L., G.L. contributed to gene analysis. All authors approved the final manuscript as submitted.
Corresponding authors
Ethics declarations
Ethical approval and consent to participate
This study was approved by the ethics committee of Shanghai Children’s Medical Center, Shanghai Jiaotong University School of Medicine (SCMCIRB-W2021044) and was performed in accordance with the Declaration of Helsinki. Informed consent for publication was obtained from all of the families of individual participants included in the study.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the journal editor.
Competing interests
The authors declare that they have no conflicts of interest related. The authors state that the material is original, has not been previously published, and has not been submitted for publication elsewhere while under consideration.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chang, G., Li, Q., Li, N. et al. Evaluating the variety of GNAS inactivation disorders and their clinical manifestations in 11 Chinese children. BMC Endocr Disord 22, 70 (2022). https://doi.org/10.1186/s12902-022-00941-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-022-00941-8