
Abstract
Background
Emerging evidences have demonstrated that gut microbiota composition is associated with pulmonary arterial hypertension (PAH). However, the underlying causality between intestinal dysbiosis and PAH remains unresolved.
Method
An analysis using the two-sample Mendelian randomization (MR) approach was conducted to examine the potential causal relationship between gut microbiota and PAH. To assess exposure data, genetic variants associated with 196 bacterial traits were extracted from the MiBioGen consortium, which included a sample size of 18,340 individuals. As for the outcomes, summary statistics for PAH were obtained from the NHGRI-EBI GWAS Catalog, which conducted a meta-analysis of four independent studies comprising a total of 11,744 samples. Causal effects were estimated employing various methods, including inverse variance weighted (IVW), MR-Egger, weighted median, weight mode and simple mode, with sensitivity analyses also being implemented with Cochran’s Q test, MR-Egger intercept test, MR-PRESSO, leave-one-out analysis, and funnel plots.
Results
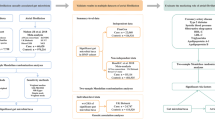
Following false discovery rate (FDR) correction, the genetically predicted genus Eubacterium fissicatena group (odds ratio (OR) 1.471, 95% confidence interval (CI) 1.178–1.837, q = 0.076) exhibited a causal association with PAH. In addition, the genus LachnospiraceaeUCG004 (OR 1.511, 95% CI 1.048–2.177) and genus RuminococcaceaeUCG002 (OR 1.407, 95% CI 1.040–1.905) showed a suggestive increased risk of PAH, while genus Eubacterium eligens group (OR 0.563, 95% CI 0.344–0.922), genus Phascolarctobacterium (OR 0.692, 95% CI 0.487–0.982), genus Erysipelatoclostridium (OR 0.757, 95% CI 0.579–0.989) and genus T–yzzerella3 (OR 0.768, 95% CI 0.624–0.945) were found to have nominal protective effect against PAH.
Conclusion
The findings from our MR study have revealed a potential causal relationship between gut microbiota and PAH. Specifically, we have identified four types of gut microbiota that exhibit a protective effect on PAH, as well as three types that have a detrimental impact on PAH, thereby offering valuable insights for future mechanistic and clinical investigations in the field of PAH.
Similar content being viewed by others
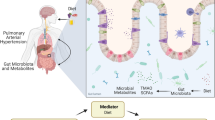
Background
Pulmonary hypertension (PH) is characterized by remodeling of the pulmonary artery, resulting in irreversible right heart failure and progressive symptoms that often lead to fatality. The extracellular matrix remodeling and fibrosis in the pulmonary vessels, which correlate with loss of compliance, have been linked to chronic perivascular inflammation and immune dysregulation [1,2,3]. Among the five subtypes of PH, group 1 is known as pulmonary arterial hypertension (PAH) [4]. It was 73.5% for patients with PAH to survive 5 years without lung transplants [5]. Patients with other subgroups of PH can receive treatment for their underlying conditions [6,7,8]. However, PAH lacks alternative therapeutic options and can only be managed through drugs that target the carbon monoxide pathway, the endothelin pathway, and the prostacyclin pathway. Despite efforts, no new therapeutic pathways have been proven effective for the treatment of PAH since 2005 [9, 10]. Moreover, treatments for PAH have proven limited in effectiveness to date, and no cure is available.
Emerging evidences suggest that the migration of gut-derived microbes and microbial products to the lungs plays a pivotal role in the pathogenesis of numerous diseases [11, 12], including PAH [13, 14]. Patients with PAH exhibit a greater prevalence of various bacteria that typically promote inflammation, as well as a decreased prevalence of certain species with anti-inflammatory properties, compared to healthy control subjects [13,14,15]. This phenomenon was similarly observed in animal experiments, and the α diversity of the gut microbiota in different ways induced a decrease in animal models compared to the normal groups [16]. Additionally, the ratio of Firmicutes to Bacteroides increased [16,17,18], which served as a sensitive biomarker of gut dysbiosis. The aforementioned findings offer substantiation for the notion that the interplay between the gut microbiota and their metabolites serves as the mediator of the intestinal lung axis.
Nevertheless, despite the distinct alterations observed in the intestinal flora of individuals with PAH and animal models in previous studies, the underlying causal connection between intestinal dysbiosis and PAH remains unresolved. Investigating this causality is of significant clinical importance, as it may contribute to understanding the lung-gut axis in PAH development and facilitate the identification of potential therapeutic targets.
In this context, Mendelian randomization (MR) studies offer an approach to address these limitations by genetically evaluating the genuine causal association between exposure and outcome [19]. This methodology effectively mitigates the influence of unobserved confounding variables, as genetic variants are randomly allocated during conception and thus independent of adaptive lifestyle factors and behaviours. This study employed the genome-wide association study (GWAS) summary statistics obtained from the MiBioGen and NHGRI-EBI GWAS Catalog to conduct a two-sample MR analysis, aiming to assess the causal relationship between gut microbiota and PAH.
Method
Study design
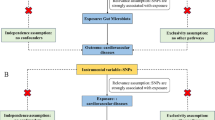
A two-sample MR study was performed to assess the causal relationship between the gut microbiota and PAH. An overview of the study description is presented in the figure below (Fig. 1). Simultaneously, adherence to three fundamental assumptions of MR design is crucial to ensure the validity of instrumental variables: 1) instrumental variables (IV), represented by genetic variations, should exhibit a significant correlation with the gut microbiota (exposure); 2) genetic variations must be independent of both known and unknown confounding factors; and 3) there should be no direct correlation between IV and PAH (outcome).

The flow chart of the study. GWAS = genome-wide association study; LD = linkage disequilibrium; MR = Mendelian randomization; MAF = minor allele frequency; MR-PRESSO = Mendelian randomization pleiotropy residual sum and outlier; SNP = single nucleotide polymorphism
GWAS data sources
The gut microbiota data utilized in this study were acquired from the international consortium MiBioGen (http://mibiogen.gcc.nl), to our knowledge, which is recognized as the largest publicly accessible sample size Genome-Wide Association Study (GWAS) of the gut microbiome. The datasets encompass the most recent comprehensive meta-analysis of genome-wide proportions, involving 18,340 individuals from 24 population-based cohorts, mostly derived from European populations (N = 13,266). Due to the diverse characteristics of age, sex ratio, and diet among cohorts, the researchers employed per-cohort and whole-study filtering methods to determine the taxa included in GWAS analyses.
The summary statistics of PAH were downloaded from the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas) on August 27, 2023 for study GCST007228 [20, 21], which conducted a meta-analysis of four independent studies comprising a total of 11,744 samples (2085 PAH cases). These studies include: 1) the UK National Institute of Health Research Bio-Resource (NIHRBR) for Rare Diseases study, which recruited between January 29, 2003 and January 4, 2017; 2) the US National Biological Sample and Data Repository for Pulmonary Arterial Hypertension/PAH Biobank (PAHB) study, and PAH cases recruited between October 3, 2012 and March 14, 2016; and 3) the Paris Pulmonary Hypertension Allele-Associated Risk cohort (PHAAR) study, all of whom were identified by the French PAH Network from 1 January 2003 to 1 April 2010. Similarly, 4) the British Heart Foundation Pulmonary Arterial Hypertension GWAS (BHFPAH) study was recruited from 3 Dec 1998 to 1 Dec 2011 (Table 1).
As the present study constitutes a reanalysis of previously published data, the acquisition of supplementary ethical approval was deemed unnecessary.
SNP selection
To establish the causal association between the gut microbiota and PAH, we employed a rigorous instrumental variable selection process: 1) Given that only a limited number of gut microbiota possessed three or more independent SNPs at the genome-wide significance threshold (P < 5 × 10−8), we employed a more lenient threshold (P < 1 × 10−5) to include additional SNPs. This approach was adopted to enhance the availability of SNPs for conducting sensitivity analyses, as in previous studies [22]. 2) The reference panel for calculating the linkage disequilibrium (LD) between SNPs consisted of European sample data from the 1000 Genomes project. Among the SNPs with r2 < 0.001 (using a clum** window size of 10 Mb) [44], thereby inducing inflammation. Intriguingly, comparable findings were also observed in hypertensive patients, with normotensive individuals displaying elevated levels of Eubacterium eligens, while essential hypertensive subjects exhibited high levels of Eubacterium fissicatena [45]. Taken together, some gut microbiota may exert SCFA-related anti-proinflammatory effects by preventing PAH initiation and development.
However, it is worth noting that not all SCFA microbial producers possess beneficial features. The families Lachnospiraceae and Ruminococcaceae possess the capacity to produce butyrate and other SCFAs through distinct biosynthetic pathways [46, 47]. In contrast, the findings from our MR analysis revealed that both the genus Lachnospiraceae UCG004 and the genus Ruminococcaceae UCG002 were suggestively associated with increased risks of inducing PAH. The abundance of the two taxa also exhibits an increase within the intestinal lumen of individuals afflicted with various diseases and elderly individuals [48,49,50,51,52]. However, members of this family have consistently demonstrated their capacity to generate favorable metabolites to the host.
However, this study is subject to certain limitations, which necessitate a more cautious interpretation of the findings. The first limitation is the inability to conduct subgroup analysis, such as assessing the severity of pulmonary arterial hypertension (PAH), due to the unavailability of individual level data. This is significant as alterations in intestinal flora among PAH patients were not found to be associated with changes in right heart function [13]. In addition, the limitation of the exposure dataset to the genus level hinders our ability to investigate the causal relationship between gut microbiota and PAH at the species level. Furthermore, the predominance of participants of European descent in genome-wide association studies restricts the generalizability of our findings to other populations. Finally, while our findings establish a causal association between specific gut microbiota and PAH, further research is needed to elucidate the underlying mechanisms.
Conclusions
The results of our MR study identified a potential causal effect of the gut microbiota and PAH. This valuable finding probably contributes to the identification of specific intestinal bacteria as biomarkers for pulmonary PAH, as well as clinical prevention and intervention of PAH through the implementation of fecal microbiota transplantation. Together, despite the presence of compelling evidence connecting gut dysbiosis to the initial development of PAH, the utilization of intestinal microbiota as a therapeutic intervention in clinical settings still requires significant advancements. It is imperative to conduct meticulous experimental investigations to establish the causative relationship between gut dysbiosis, altered gut microbiome, and the pathogenesis of PAH prior to considering the modulation of gut microbiota as a viable therapeutic approach for treating PAH.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- BHFPAH:
-
British Heart Foundation Pulmonary Arterial Hypertension study
- CI:
-
Confidence interval
- FDR:
-
False discovery rate
- GPRs:
-
G-protein-coupled receptors
- GWAS:
-
Genome-wide association studies
- HDAC:
-
Histone deacetylase
- IV:
-
Instrumental variables
- IVW:
-
Inverse variance weighted
- LD:
-
Linkage disequilibrium
- mPAP:
-
Mean pulmonary artery pressure
- MAF:
-
Minor allele frequency
- MR:
-
Mendelian randomization
- MR-PRESSO:
-
MR pleiotropy residual sum and outlier
- NIHRBR:
-
National Institute for Health Research BioResource study
- OR:
-
Odds ratio
- PAH:
-
Pulmonary arterial hypertension
- PAHB:
-
PAH Biobank study
- PCWP:
-
pulmonary capillary wedge pressure
- PH:
-
Pulmonary hypertension
- PHAAR:
-
Pulmonary Hypertension Allele-Associated Risk study
- PVR:
-
Pulmonary vascular resistance
- SCFA:
-
Short-chain fatty acid
- SNP:
-
Single nucleotide polymorphisms
References
Goncharova EA, Kudryashova TV, Maroli G, Pullamsetti SS. Matrix Metalloproteinase-8 in pulmonary hypertension: the sheep in the Wolf's skin? Am J Respir Crit Care Med. 2021;204(12):1361–3. https://doi.org/10.1164/rccm.202109-2144ED.
Johnson S, Sommer N, Cox-Flaherty K, Weissmann N, Ventetuolo CE, et al. Pulmonary hypertension: a contemporary review. Am J Respir Crit Care Med. 2023;208(5):528–48. https://doi.org/10.1164/rccm.202302-0327SO.
Lian G, You J, Lin W, Gao G, Xu C, Wang H, et al. Bioinformatics analysis of the immune cell infiltration characteristics and correlation with crucial diagnostic markers in pulmonary arterial hypertension. BMC Pulm Med. 2023;23(1):300. https://doi.org/10.1186/s12890-023-02584-4.
Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913. https://doi.org/10.1183/13993003.01913-2018.
Galiè N, Channick RN, Frantz RP, Grünig E, **g ZC, Moiseeva O, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889. https://doi.org/10.1183/13993003.01889-2018.
Ruaro B, Salton F, Baratella E, Confalonieri P, Geri P, Pozzan R, et al. An overview of different techniques for improving the treatment of pulmonary hypertension secondary in systemic sclerosis patients. Diagnostics (Basel). 2022;12(3):616. https://doi.org/10.3390/diagnostics12030616.
Balsa A, Adão R, Brás-Silva C. Therapeutic approaches in pulmonary arterial hypertension with beneficial effects on right ventricular function-preclinical studies. Int J Mol Sci. 2023;24(21):15539. https://doi.org/10.3390/ijms242115539.
Wang Z, Li X, Li M, Peng J, Zhang H. The efficacy of the treat-repair-treat strategy for severe pulmonary arterial hypertension associated with congenital heart disease: a meta-analysis. BMC Cardiovasc Disord. 2023;23(1):569. https://doi.org/10.1186/s12872-023-03606-z.
Humbert M, Guignabert C, Bonnet S, Dorfmüller P, Klinger JR, Nicolls MR, et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1):1801887. https://doi.org/10.1183/13993003.01887-2018.
Condon DF, Agarwal S, Chakraborty A, Auer N, Vazquez R, Patel H, et al. Novel mechanisms targeted by drug trials in pulmonary arterial hypertension. Chest. 2022;161(4):1060–72. https://doi.org/10.1016/j.chest.2021.10.010.
Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16(1):35–56. https://doi.org/10.1038/s41575-018-0061-2.
Lynch SV, Pedersen O. The human intestinal microbiome in health and disease. N Engl J Med. 2016;375(24):2369–79. https://doi.org/10.1056/NEJMra1600266.
Moutsoglou DM, Tatah J, Prisco SZ, Prins KW, Staley C, Lopez S, et al. Pulmonary arterial hypertension patients have a Proinflammatory gut microbiome and altered circulating microbial metabolites. Am J Respir Crit Care Med. 2023;207(6):740–56. https://doi.org/10.1164/rccm.202203-0490OC.
Kim S, Rigatto K, Gazzana MB, Knorst MM, Richards EM, Pepine CJ, et al. Altered gut microbiome profile in patients with pulmonary arterial hypertension. Hypertension. 2020;75(4):1063–71. https://doi.org/10.1161/HYPERTENSIONAHA.119.14294.
Huang Y, Lin F, Tang R, Bao C, Zhou Q, Ye K, et al. Gut microbial metabolite trimethylamine N-oxide aggravates pulmonary hypertension. Am J Respir Cell Mol Biol. 2022;66(4):452–60. https://doi.org/10.1165/rcmb.2021-0414OC.
Hong W, Mo Q, Wang L, Peng F, Zhou Y, Zou W, et al. Changes in the gut microbiome and metabolome in a rat model of pulmonary arterial hypertension. Bioengineered. 2021;12(1):5173–83. https://doi.org/10.1080/21655979.2021.1952365.
Sharma RK, Oliveira AC, Yang T, Kim S, Zubcevic J, Aquino V, et al. Pulmonary arterial hypertension-associated changes in gut pathology and microbiota. ERJ Open Res. 2020;6(3):00253–2019. https://doi.org/10.1183/23120541.00253-2019.
Callejo M, Barberá JA, Duarte J, Perez-Vizcaino F. Impact of nutrition on pulmonary arterial hypertension. Nutrients. 2020;12(1):169. https://doi.org/10.3390/nu12010169.
Smith GD, Ebrahim S. 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. https://doi.org/10.1093/ije/dyg070.
Sollis E, Mosaku A, Abid A, Buniello A, Cerezo M, Gil L, et al. The NHGRI-EBI GWAS catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51(D1):D977–85. https://doi.org/10.1093/nar/gkac1010.
Rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, et al. Genetic determinants of risk in pulmonary arterial hypertension: international genome-wide association studies and meta-analysis. Lancet Respir Med. 2019;7(3):227–38. https://doi.org/10.1016/S2213-2600(18)30409-0.
Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Võsa U, Mujagic Z, et al. Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases. Nat Genet. 2019;51(4):600–5. https://doi.org/10.1038/s41588-019-0350-x.
Clarke L, Zheng-Bradley X, Smith R, Kulesha E, **ao C, Toneva I, et al. 1000 genomes project consortium. The 1000 genomes project: data management and community access. Nat Methods. 2012;9(5):459–62. https://doi.org/10.1038/nmeth.1974.
Slob EAW, Burgess S. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol. 2020;44(4):313–29. https://doi.org/10.1002/gepi.22295.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44(2):512–25. https://doi.org/10.1093/ije/dyv080.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98. https://doi.org/10.1093/ije/dyx102.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat Genet. 2018;50(5):693–8. https://doi.org/10.1038/s41588-018-0099-7.
Hemani G, Tilling K, Davey SG. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13(11):e1007081. https://doi.org/10.1371/journal.pgen.1007081.
Marsh LM, Jandl K, Grünig G, Foris V, Bashir M, Ghanim B, Klepetko W, Olschewski H, Olschewski A, Kwapiszewska G. The inflammatory cell landscape in the lungs of patients with idiopathic pulmonary arterial hypertension. Eur Respir J. 2018;51(1):1701214. https://doi.org/10.1183/13993003.01214-2017.
Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54(1 Suppl):S10–9. https://doi.org/10.1016/j.jacc.2009.04.006.
Huertas A, Tu L, Humbert M, Guignabert C. Chronic inflammation within the vascular wall in pulmonary arterial hypertension: more than a spectator. Cardiovasc Res. 2020;116(5):885–93. https://doi.org/10.1093/cvr/cvz308.
Lucas S, Omata Y, Hofmann J, Böttcher M, Iljazovic A, Sarter K, et al. Short-chain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat Commun. 2018;9(1):55. https://doi.org/10.1038/s41467-017-02490-4.
Kayama H, Okumura R, Takeda K. Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu Rev Immunol. 2020;38:23–48. https://doi.org/10.1146/annurev-immunol-070119-115104.
Augustin A, Guennec AL, Umamahesan C, Kendler-Rhodes A, Tucker RM, Chekmeneva E, et al. Faecal metabolite deficit, gut inflammation and diet in Parkinson's disease: integrative analysis indicates inflammatory response syndrome. Clin Transl Med. 2023;13(1):e1152. https://doi.org/10.1002/ctm2.1152.
Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–6. https://doi.org/10.1038/nature08530.
Vinolo MA, Ferguson GJ, Kulkarni S, Damoulakis G, Anderson K, Bohlooly-Y M, et al. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS One. 2011;6(6):e21205. https://doi.org/10.1371/journal.pone.0021205.
Nicolas GR, Chang PV. Deciphering the chemical lexicon of host-gut microbiota interactions. Trends Pharmacol Sci. 2019;40(6):430–45. https://doi.org/10.1016/j.tips.2019.04.006.
Yan J, Pan Y, Shao W, Wang C, Wang R, He Y, et al. Beneficial effect of the short-chain fatty acid propionate on vascular calcification through intestinal microbiota remodelling. Microbiome. 2022;10(1):195. https://doi.org/10.1186/s40168-022-01390-0.
Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–39. https://doi.org/10.1016/j.immuni.2013.12.007.
Kaisar MMM, Pelgrom LR, van der Ham AJ, Yazdanbakhsh M, Everts B. Butyrate conditions human dendritic cells to prime type 1 regulatory T cells via both histone deacetylase inhibition and G protein-coupled receptor 109A signaling. Front Immunol. 2017;8:1429. https://doi.org/10.3389/fimmu.2017.01429.
Lu H, Xu X, Fu D, Gu Y, Fan R, Yi H, et al. Butyrate-producing Eubacterium rectale suppresses lymphomagenesis by alleviating the TNF-induced TLR4/MyD88/NF-κB axis. Cell Host Microbe. 2022;30(8):1139–1150.e7. https://doi.org/10.1016/j.chom.2022.07.003.
Chung WSF, Meijerink M, Zeuner B, Holck J, Louis P, Meyer AS, et al. Prebiotic potential of pectin and pectic oligosaccharides to promote anti-inflammatory commensal bacteria in the human colon. FEMS Microbiol Ecol. 2017;93(11) https://doi.org/10.1093/femsec/fix127.
Karoor V, Strassheim D, Sullivan T, Verin A, Umapathy NS, Dempsey EC, et al. The short-chain fatty acid butyrate attenuates pulmonary vascular remodeling and inflammation in hypoxia-induced pulmonary hypertension. Int J Mol Sci. 2021;22(18):9916. https://doi.org/10.3390/ijms22189916.
Wang B, Kong Q, Li X, Zhao J, Zhang H, Chen W, et al. A high-fat diet increases gut microbiota biodiversity and energy expenditure due to nutrient difference. Nutrients. 2020;12(10):3197. https://doi.org/10.3390/nu12103197.
Nakai M, Ribeiro RV, Stevens BR, Gill P, Muralitharan RR, Yiallourou S, et al. Essential hypertension is associated with changes in gut microbial metabolic pathways: a multisite analysis of ambulatory blood pressure. Hypertension. 2021;78(3):804–15. https://doi.org/10.1161/HYPERTENSIONAHA.121.17288.
Flint HJ. Gut microbial metabolites in health and disease. Gut Microbes. 2016;7(3):187–8. https://doi.org/10.1080/19490976.2016.1182295.
Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol. 2014;12(10):661–72. https://doi.org/10.1038/nrmicro3344.
Maffei VJ, Kim S, Blanchard E 4th, Luo M, Jazwinski SM, Taylor CM, et al. Biological aging and the human gut microbiota. J Gerontol A Biol Sci Med Sci. 2017;72(11):1474–82. https://doi.org/10.1093/gerona/glx042.
Teng Y, Mu J, Xu F, Zhang X, Sriwastva MK, Liu QM, et al. Gut bacterial isoamylamine promotes age-related cognitive dysfunction by promoting microglial cell death. Cell Host Microbe. 2022;30(7):944–960.e8. https://doi.org/10.1016/j.chom.2022.05.005.
Jiang Z, Zhuo LB, He Y, Fu Y, Shen L, Xu F, et al. The gut microbiota-bile acid axis links the positive association between chronic insomnia and cardiometabolic diseases. Nat Commun. 2022;13(1):3002. https://doi.org/10.1038/s41467-022-30712-x.
Zhang X, Yu D, Wu D, Gao X, Shao F, Zhao M, et al. Tissue-resident Lachnospiraceae family bacteria protect against colorectal carcinogenesis by promoting tumor immune surveillance. Cell Host Microbe. 2023;31(3):418–432.e8. https://doi.org/10.1016/j.chom.2023.01.013.
Ghosh TS, Rampelli S, Jeffery IB, Santoro A, Neto M, Capri M, et al. Mediterranean diet intervention alters the gut microbiome in older people reducing frailty and improving health status: the NU-AGE 1-year dietary intervention across five European countries. Gut. 2020;69(7):1218–28. https://doi.org/10.1136/gutjnl-2019-319654.
Acknowledgements
We gratefully acknowledge the authors and participants of all GWAS from which we used summary statistics data.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81870049, 82170060).
Author information
Authors and Affiliations
Contributions
YY and YH designed the study and drafted the manuscript. SL, MY, YY, CZ participated in data collection and statistical analyses. All authors agreed on the content of the manuscript, reviewed manuscript, and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, Y., Li, S., Yan, M. et al. Genetically determined gut microbiota associates with pulmonary arterial hypertension: a Mendelian randomization study. BMC Pulm Med 24, 235 (2024). https://doi.org/10.1186/s12890-024-02877-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-024-02877-2