
Abstract
Background
Bacteria in lung play an important role in sustaining lung health. Understanding the characteristics of bacteriomes in lesions of pulmonary tuberculosis (TB) patients, who excrete Mycobacterium tuberculosis (MTB), is important for TB prevention and effective treatment.
Methods
In this study, bacteriomes in lesions from TB patients excreting bacteria (TB-E) and those from TB patients not excreting bacteria (TB-NE) with matched normal lung tissues (NT) were compared by 16S rRNA sequencing. Bacterial MetaCyc functions in TB lesions were also predicted by PICRUSt2 tool.
Results
Alpha diversity of bacteria, including Chao 1 and Shannon indexes, for TB-E was significantly higher than those in TB-NE and NT; while for TB-NE group, Chao 1 index was higher than that in NT group. Predominant phyla in TB lesions and NT were Proteobacteria, Actinobacteria, Firmicutes, and Bacteroidetes, but analysis of similarity (ANOSIM, p < 0.001) revealed significantly different bacterial compositions among TB-E, TB-NE and NT samples. As for bacteriomes in TB lesions, a strong association (ANOSIM, p < 0.001) was observed with the status of MTB excretion. Indicator genera identified in TB-E and TB-NE demonstrated distinctive micro-ecological environments of TB lesions from patients with different clinical manifestations. Co-occurrence analysis revealed a densely-linked bacterial community in TB-NE compared to that in TB-E. MetaCyc functions responsible for menaquinone synthesis and chorismate metabolism that could potentially impact the persistent-state and nutrient metabolism of MTB were enriched in TB-E samples. While in TB-NE samples, enrichment of bacterial MetaCyc function responsible for heme b synthesis might contribute to TB pathology through ferroptosis.
Conclusion
Bacteriomes and their MetaCyc functions in TB lesions are elucidated, and they are associated with status of MTB excretion among pulmonary TB patients. These results serve as a basis for designing novel strategies for preventing and treating pulmonary TB disease.
Similar content being viewed by others

Background
Pulmonary tuberculosis (TB), caused by Mycobacterium tuberculosis (MTB) infection, is transmissible through the airborne route [1]. Only patients with sputum culture-positive are considered infectious, and they can threaten public health. Although antibiotic treatment is the fundamental therapeutic regimen for pulmonary TB according to the World Health Organization (WHO) [2], the conversion time of sputum culture varies greatly between individuals [3].
Organized granuloma is a typical lesion of pulmonary TB patients. TB lesions are reservoirs for MTB survival in the lung. Physiological and biochemical conditions of TB lesions, such as penetration of antibiotic drugs, oxygen tension, acidic pH, and lipid abundance, would impact MTB’s status of persistence, replication and transmission [4]. When the center of the granuloma liquefies and ruptures, MTB bacilli become extracellular, and it can be released into the air through coughing, talking, or sneezing, which directly results in sputum culture-positive outcomes. Negative conversion of sputum culture is a crucial indicator to monitor patient’s prognosis during antituberculosis treatment. Many non-antibiotic factors, including history of smoking [5], chest cavity [3], body mass index (BMI) [6], etc., have been reported to be significantly associated with the time of sputum culture conversion. However, it is the status of MTB excretion in TB lesions that ultimately determines the conversion of sputum culture results.
Pulmonary inherent bacteria have been proven essential both in healthy and diseased status of the lung [7, 8]. Furthermore, it can promote the host’s resistance to MTB colonization through regulating mucosal-associated invariant T (MAIT) cells in the lung of mice model [9]. Besides the physiological and biochemical impacts of TB lesions on MTB colonization [4], lung bacteria may be another important factor that can affect the growth behavior of MTB. Many studies have investigated the lung bacteria of pulmonary TB patients, but they were carried out using sputum [10,11,12] or bronchoalveolar lavage fluid (BLF) samples [13, 14]. However, microbiota in sputum and BLF samples cannot exactly represent the entire inhabitants in lung, specifically in TB lesions. So far, the characteristics of bacteriomes in TB lesions have not been clearly investigated yet. Taken together, whether the inherent bacteria in TB lesions contribute to the conversion of MTB excretion status is poorly understood.
In the current study, TB lesions and normal lung samples were respectively collected from pulmonary TB and lung cancer patients, who have undergone surgical treatment. Sputum culture results of MTB for all enrolled pulmonary TB patients were also collected. We explored the characteristics of bacterial composition in TB lesions and normal tissues using 16S rRNA sequencing method to 1) compare the distinctive features of bacterial profiles in TB lesions and normal lung tissues, and 2) analyze the association between pulmonary bacteria and MTB excretion status in TB patients. Metabolic functions of bacteria in TB lesions were also predicted using the phylogenetic investigation of communities by reconstruction of unobserved states 2 (PICRUSt2) tool to reveal the correlations between predicted functions and status of TB lesions.
Methods
Clinical characterization of the participants
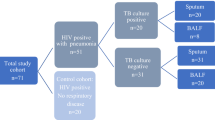
Formalin fixed and paraffin embedded (FFPE) TB lesions and normal lung tissues (NT) from pulmonary TB patients and lung cancer patients, who have undergone surgical treatment, were obtained in the Department of Pathology, Bei**g Chest Hospital. The clinical indications for biopsy in patients with tuberculosis were that the anti-tuberculosis drug treatment was ineffective and the lesions were relatively localized. The biopsy samples used in this study were obtained from lung through surgical operation for pathological morphological and molecular diagnosis of TB disease. The standard for the diagnosis of pulmonary TB disease was to meet any of the followings: 1) mycobacterial culture for sputum was positive; or 2) TB-PCR of IS6110 fragment for TB lesion was positive. Patients whose sputum culture results were positive were classified as TB patients excreting bacteria (TB-E) or TB patients not excreting bacteria (TB-NE) if their sputum culture results were negative but TB-PCR for TB lesions were positive. Totally, 82 pulmonary TB patients were obtained, including 40 TB-E and 42 TB-NE cases, respectively. Additionally, 18 lung cancer patients, whose NT samples > 2 cm distal to tumor lesions, were enrolled as bacterial baseline. All participants had not taken any medications for human immunodeficiency disease before samples collection.
DNA extraction and processing
According to the hematoxylin–eosin (HE) staining results, the normal lung tissues surrounding the TB lesions were removed from the FFPE samples, and 0.5–1.0 cm2 lesion areas were retained. These HE-stained lesion areas showed necrotic or non-necrotic granulomatous inflammation with visible epithelial cells, chronic inflammatory cells, and/or Langhans giant cells surrounded by fibrous connective tissue. NT samples with 0.5–1.0 cm2 areas were selected for this study, and HE-stained NT samples showed normal lung lobule structure. Twenty Sects. (4 μm) of TB lesions or NT samples were used to extract DNA using QIAamp DNA FFPE Tissue Kit 56,404 (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. The concentration of the DNA was measured using a NanoDrop 2000 spectrometer (Thermo Fisher Scientific, Waltham, United States). The integrity of DNA was evaluated by 1% agarose gel electrophoresis.
Amplification of 16S rRNA V4 hypervariable region was carried out with primer sets, 515F (5’-GTGCCAGCMGCCGCGGTAA-3’) and 806R (5’-GGACTACHVGGGTWTCTAAT-3’), with barcodes at the 5’ end of the forward primers. The PCR reaction system was 50 μL, with 1.5 μL primer sets (10 μmol/L), 25 μL 2 × KAPA HiFi HotStart ReadyMix (Kapa Biosystems, MA, United States) and 23 μL extracted FFPE DNA. The PCR protocol included: initial denaturation at 95 ℃ for 3 min, 30 cycles of denaturation (20 s at 98 ℃), annealing (15 s at 55 ℃) and extension (15 s at 72 ℃), and a final extension at 72 ℃ for 10 min. The PCR reaction was triplicated for each sample, and the three PCR products were pooled together and purified through 2% agarose gel electrophoresis. The concentration of purified PCR products was quantified using a Qubit 4.0 fluorometer (Life ABI, Waltham, United States) before DNA library construction. Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, California) was used to confirm fragment size and distribution of the constructed DNA library. The library was barcode indexed with the NEB Next Ultra DNA Library Prep Kit for Illumina (New England Biolabs, United States), and sequenced on Illumina MiSeq PE 250 machine.
16S rRNA gene analysis
After sequence demultiplex, the raw data were analyzed using QIIME v1.9 [15]. The sequences were quality trimmed and filtered (fastq_maxee = 0.5) using QIIME default parameters, and qualified sequences were clustered into OTUs (operational taxonomic unit) by the closed-reference OTU picking methods at 97% sequence similarity using USEARCH v7.0.1090 [16]. The filtered OTUs with the number of sequences < 0.005% of the total were discarded [17]. Greengenes database (August 2013 release) was referred to annotate bacterial taxa. OTUs that mapped to the mitochondria and chloroplasts were discarded. All samples were then rarefied to the same sequence number, according to the number in the sample that yielded the fewest sequences. The final rarefied OTU table, providing the abundances of bacterial taxa in each sample, was used for downstream analyses. QIIME was also used to create α-diversity indexes, including Chao1 richness, Shannon, and Good’s coverage, as well as β-diversity index (Bray–Curtis distance metrics). PICRUSt2 was used to generate a MetaCyc metabolic pathway to predict the functions of 16S rRNA sequences in each sample [18].
Statistical analysis
Bacterial variety among different pulmonary tissue categories was visualized by principal coordinates analysis (PCoA), based on Bray–Curtis distance using the “ggplot2” package for R [19]. To statistically verify the bacterial clustering in the PCoA analysis, the analysis of similarity (ANOSIM) algorithm was utilized. Heatmap with K-means clustering analysis was made using “pheatmap” package for R [20]. The Venn diagram was drawn using the “VennDiagram” package [21]. Bacterial indicators in different categories were predicted using “indicspecies” packages, with n = 999 random permutations. Analysis of variance (ANOVO) and Tukey’s test together were used to calculate statistical differences of α-diversity indexes between different tissue categories. Two-sample proportion z test was used to calculate the statistical differences of bacterial species and MetaCyc functions between two categories. These analyses above were performed in the R software program (v.3.2.1; http://www.r-project.org).
Results
Clinical characteristics of pulmonary TB patients
Detailed clinical and demographical characteristics of pulmonary TB patients are listed in Table 1. Eighty two pulmonary TB lesions from 82 patients were obtained, including 40 TB-E and 42 TB-NE cases, respectively. The number of patients who had cough, fever, night sweats, weight loss, or cavitation on chest CT in the TB-E group showed no statistical differences compared to those in the TB-NE group. Patients in the TB-NE group were 9.4 years younger than those in the TB-E group (p = 0.005). The proportion of patients with hemoptysis symptoms in the TB-E group was significantly lower than that in the TB-NE group (p = 0.003). A major proportions of TB patients in TB-E (72.5%, 29/40) and TB-NE (65.9%, 27/42) groups have received standard HREZ regimens (ie isoniazid, rifampicin, pyrazinamide and ethambutol). Also, the numbers of patients receiving standard HRZE regime showed no difference (p = 0.684) between the two groups. It should be noted that IS6110 amplifications of the 82 TB lesion tissues were all positive, though acid fast bacilli (AFB) positive ratios of TB lesion tissues from TB-E and TB-NE patients were 67.5% (27/40) and 71.4% (30/42). Also, we collected 18 NT samples from 18 lung cancer patients to be used as the bacterial baseline.
Distinct bacterial compositions among TB-E, TB-NE and NT samples
A data set of 16S rRNA gene fragments with 5,670,646 valid sequences was obtained after quality control and filtration of DNA fragments from mitochondria and chloroplast. Sequence numbers for each sample ranged from 33,658 to 65,701. To avoid the bacterial discrepancy caused by different sequencing depths, all sample sequences were rarefied to 33,658 before further analysis. Based on the 97% similarity threshold, 1,205 OTUs were clustered from these sequences. Venn diagram showed that 63.40% (764/1,205) of OTUs were shared by all the samples (Fig. 1A). The proportions of specific OTUs in the TB-E group (5.81%, 70/1,205) and the TB-NE group (5.31%, 64/1,205) were similar, but both much higher than that in the NT group (0.08%, 1/1,205) (Fig. 1A). The single specific OTU in NT group was an uncultured bacterium belonged to genus Rubellimicrobium. Proteobacteria, Actinobacteria, Firmicutes, Bacteroidetes, and Cyanobacteria were the major phyla observed in human lung tissues, which accounted for 94.4% of the sequences in total (Fig. 1B). Additionally, the archaeal phylum Euryarchaeota was also detected, and it was more predominantly in TB-E samples (100%, 40/40) than that in TB-NE samples (45.2%, 19/42). Phylum Euryarchaeota was rarely reported in previous TB microbiome studies using sputum or BAL samples. Within the TB-NE group, Proteobacteria accounted for a higher sequence number (p < 0.001), while Firmicutes accounted for a lower sequence number (p = 0.006) when compared to the TB-E samples. A larger proportion of phylum Cyanobacteria were observed in TB-E (p = 0.003) and TB-NE (p = 0.014) groups than that in NT samples. In genus level, predominant bacteria in TB lesions and NT samples were Pseudomonas, Bacteroides, Mycobacterium, Sphingomonas, Ottowia and Cutibacterium. However, NT group was enriched with Pseudomonas and Bacteroides; while TB-E and TB-NE were both enriched with Mycobacterium.

TB-E, TB-NE and normal lung tissues (NT) have distinct bacterial diversities. A Venn diagram of the exclusive and shared OTUs. B Average relative abundances of phylum-level taxa showing the distribution of bacterial compositions. C and D Box plot of Chao1 and Shannon index of the three tissue categories. E Principal coordinate analysis (PCoA) based on Bray–Curtis dissimilarity displaying the bacterial variations. Statistical differences are calculated using the analysis of similarity (ANOSIM) algorithm. The dotted circles indicate the 95% confidence intervals. TB-E: Lesions from TB patients excreting bacteria; TB-NE: Lesions from TB patients not excreting bacteria
Alpha diversity of pulmonary bacteriomes was evaluated by Chao 1 and Shannon indices (Fig. 1C-D). Chao 1 and Shannon indices of TB-E were both significantly higher than those of TB-NE and NT groups (ANOVA, p < 0.05). When comparing the alpha diversity of the TB-NE group with that of the NT samples, Chao 1 index was statistically higher (p = 0.028), but Shannon index showed no significant difference (p = 0.674). Principal coordinates analysis (PCoA) demonstrated that bacterial compositions distinctly clustered in line with the three different tissue categories (Fig. 1E). This separation was further confirmed by analysis of similarity (ANOSIM) (p < 0.001). These results indicate that bacterial composition in TB lesions is significantly different from that in normal lung tissue, and is strongly associated with the MTB excretion status.
Totally, 49 OTUs assigned to genera Mycobacterium were defined from bacterial 16S rRNA sequences. Mycobacterium accounted for a proportion of 0–13.94% of the total 16S rRNA sequences in TB lesions, but their relative abundance showed no significant difference (p = 0.186) between TB-E and TB-NE groups.
Indicator genera can reflect the distinctive microenvironments in various habitats. Indicator genera in TB lesions and NT samples were identified by IndVal analysis (Fig. 2). With an indicator value > 0.70 and p < 0.05, there were respectively 16, 12 and 1 indicator genera in TB-E, TB-NE and NT samples. Indicator genera in TB lesions belonged to Actinobacteria, Bacteroidetes, Deinococcus-Thermus, Euryarchaeota, Firmicutes, Fusobacteria and Proteobacteria. Significantly, 66.7% (8/12) indicator genera in TB-NE group were Proteobacteria. An uncultured archaea affiliated to Ferroplasmaceae was also observed in TB-E group as an indicator genus. The single indicator genus in NT samples was Pseudomonas, which was not the specific taxa Rubellimicrobium described above.

IndVal analysis showing indicator genera in TB-E and TB-NE samples. Genera taxa with an indicator value > 0.70 and p < 0.05 are identified as indicators
Co-occurrence analysis of bacterial interactions in TB lesions
The specific bacterial co-occurring networks, including strong (Spearman’s ρ > 0.6) and significant (p < 0.01) correlations, were generated in the TB-E and the TB-NE samples to explore how bacteria were potentially connected with each other (Fig. 3). Nodes in the network were assigned to 10 bacterial phyla, among which two phyla (Proteobacteria and Actinobacteria) were widely distributed, together accounting for 56.12% of all nodes. However, no taxa showed statistical correlation with the genera Mycobacterium in both TB-E and TB-NE groups. Topological parameters were further calculated to describe the complex pattern of bacterial inter-relationships in TB-E and TB-NE groups. The number of nodes, edges, clustering coefficient, average path length, network diameter and degree in TB-E group were respectively 142, 136, 0.505, 2.401, 6 and 1.915, which were all lower than those in the TB-NE group (Table 2). The edge and node numbers in TB-E group were less than that in TB-NE group, for which the average path length was also lower, suggesting that bacteria in MTB non excreting lesions were less connected but those correlated with each other were quite closely connected. The smaller average path length of network for TB-E group indicated a “small-world” property, which was believed to have a more effective and close bacterial connections to response quickly to external perturbations [22].

Network of co-occurring genera showing the bacterial interactions in TB-E (A) and TB-NE (B). Each connection indicates a strong (Spearman’s ρ > 0.6) and significant (p < 0.01) correlation. The size of each node is proportional to the number of connections, and the edge thickness is proportional to the weight of each correlation
Bacterial MetaCyc functions identified in TB lesions
Totally, 449 bacterial MetaCyc functions of pulmonary bacteria were predicted using the PICRUSt2 tool based on 16S rRNA sequencing data from TB lesions (Fig. 4A, Supplemental Table 1), mainly including energy metabolism, environmental and information processing, genetic information processing, and cellular processing. These bacterial MetaCyc functions were responsible for maintaining the basic physiological activities and they were widely distributed in taxonomically distinct bacteria. Therefore, we mainly focused on 55 bacterial MetaCyc pathways that were statistically different between TB-E and TB-NE samples (Fig. 4B, Supplemental Table 2).

Bacterial metabolic functions predicted by PICRUSt in TB lesions. A Heatmap demonstrating K-means clustering of 449 metabolic pathways across TB-E and TB-NE samples. B Volcano plots showing bacterial metabolic pathways that differ in TB-E compared with TB-NE. C Volcano plot demonstrating the genera with striking differences between TB-E and TB-NE sample types. A two-sample t test was used to calculate p values in (B) and (C)
Forty-one bacterial MetaCyc pathways with statistically richer levels in TB-E samples included menaquinone biosynthesis, chorismate metabolism, penicillin resistance, hexitol degradation, nucleic acid processing etc. In TB-NE samples, 14 bacterial MetaCyc pathways that were responsible for vanillin and vanillate degradation, catechol degradation, D-galacturonate degradation, heme b biosynthesis, adenosylcobamide biosynthesis etc., were significantly enriched. Ten genera were markedly different between the two groups (Fig. 4C), but they could not explain the discrepancy of bacterial MetaCyc functions described in Fig. 4B and Table S2. This is reasonable because bacterial composition is generally decoupled from bacterial metabolic functions, and each single metabolic function is usually performed by multiple coexisting, taxonomically distinct bacteria [23]. Additionally, most bacterial taxa in TB lesions were uncultured or unidentified, and detailed information about their metabolic functions is still unknown.
Discussion
Lung bacteriomes play an important role in TB pathophysiological processes and clinical manifestations, but very few studies have investigated the bacteriomes in lower respiratory tract using lung tissues. Different from previous investigations on pulmonary bacteria in TB patients by using sputum or BAL samples [11,12,13,14, 24], we collected lung tissues to reflect the characteristics of lung bacteriomes. This study initially explored the characteristics of bacteriomes in TB lesions and normal lung tissues, and evaluated its association with MTB excretion status.
Microenvironmental conditions in the human lung, such as nutrient availability, oxygen tension, activation of inflammatory cells, and host epithelial cell interactions, will change greatly during the formation of granuloma lesions [7]. TB lesions provide new ecological niches for bacteria, resulting in a new balance of bacterial immigration, elimination, and bacterial relative production rates [25]. TB lesions are places where MTB grows and host anti-TB immune responses occur. Bacteria in TB lesions can directly interact with MTB and participate in anti-TB immune responses [9]. Dominant phyla, with relative abundance from high to low, observed in the TB lesions were successively Proteobacteria, Actinobacteria, Firmicutes, Bacteroidetes, and Cyanobacteria. Ticlla and her colleagues have reported Firmicutes, Proteobacteria, Bacteroidetes, Actinobacteria and Fusobacteria as the major phyla order in sputum samples [12]. This difference is reasonable because microbes from the upper respiratory tract and the oral cavity can contaminate sputum samples inevitably during the expectoration process, which will bias the microbial profile in sputum. In BAL samples, the relative abundance of bacteria from high to low is Actinobacteria, Firmicutes, Proteobacteria, Bacteroidetes and Fusobacteria [13], which is different from our study. Bacteria load in BAL is much lower than that in lung tissues [26], which can also lead to contamination easily during BAL sampling. As for bacterial diversity, Chao 1 and Shannon indexes in TB lesions were both higher than those in NT samples. However, bacterial Chao 1 index in TB sputum samples is higher than that in normal controls, while the Shannon index is inverse [11]. In BAL, the Shannon index in TB group is higher than that in healthy group, while the Chao 1 index is inverse [27]. These results provided a new insight of microbial profile and diversity in the lower respiratory tract using lung tissues, instead of sputum and BAL samples.
Clinical symptoms and bacterial communities in the lower respiratory tract of inter-individual TB patient vary largely [28,29,30]. We observed a strong correlation of bacterial profiles in TB lesions with the status of MTB excretion among pulmonary TB patients. Interestingly, sputum microbial composition and diversity were also reported to be associated with local lung environment indicated by abnormal chest x-ray findings [12]. Specific OTUs and indicator genera detected both suggested different micro-ecological environments of TB lesions. Co-occurrence patterns of bacteria in TB-E samples showed a “small-world” property compared to TB-NE group, indicating that bacterial community in TB-E were closely connected with each other. Bacterial composition is mainly determined by the physiological and chemical environments of TB lesions [23]. When the status of TB lesions changes from MTB excretion to non-excretion, the newly formed ecological niches in TB lesions not only selectively harbor a variety of taxonomically distinct inhaled bacteria but also change the relative reproduction rates of inherent bacterial members [7, 31]. Additionally, extremely rare taxa (< 0.1% proportion) can become dominant or reach a much higher abundance in new condition [32, 33]. These results collectively revealed a strong association of lung bacteriomes in TB lesions with local lung environment and sputum culture results.
Revealing characteristics of bacterial composition in TB lesions is important to enrich our knowledge of bacterial resources in human host. Bacterial members generally exhibit complex interactions and metabolic exchanges with each other ubiquitously [34]. Despite the bacterial diversity, bacterial functions also should not be overlooked. However, microbial functional profiles are decoupled from taxonomic composition, and specific bacterial functions are strongly controlled by the external environment [23]. Broadly distributed bacterial functions identified in TB lesions, such as energy metabolism, environmental and information processing, genetic information processing, and cellular processing, were strongly conserved in most bacterial taxa, making pulmonary micro-ecological systems more resistant to changes in taxonomic composition and diversity [35, 36]. However, multiple MetaCyc functions were also enriched in TB-E and TB-NE samples, respectively. We mainly emphasized those bacterial functions with statistically different enrichments between TB-E and TB-NE samples.
In TB-E samples, bacterial MetaCyc functions responsible for menaquinone biosynthesis and chorismate biosynthesis that were significantly enriched (Supplemental Fig. 1) have been reported to impact the survival of MTB bacillus [37,38,39,40]. Menaquinone is a small molecule and possesses the capacity to signal redox status, which makes it an essential factor in triggering the persistence of MTB [37]. Menaquinone biosynthesis is important to generate energy to sustain the growth of persistent state-like MTB [38]. In TB-E lesions, enrichment of menaquinone biosynthesis in bacteria can promote MTB into the persistent state, which can lead to negative sputum culture results. The second is MetaCyc pathway responsible for chorismate biosynthesis. After the formation of chorismate, a branching of shikimate biosynthetic pathway occurs. Shikimate pathway enzymes are essential for the biosynthesis of amino acids, phenylalanine and tyrosine in bacteria, fungi, algae, and plants [39]. The enrichment of chorismate biosynthesis reflects a stronger metabolic activity of organisms, including MTB bacillus, to scavenge for nutrients from the human host in TB lesions. Chorismate has also been considered as an attractive target for develo** antibiotic agents for MTB bacillus [40]. It also should be noted that the two bacterial functions can be predicted both from mycobacteria and co-existing genera based on the MetaCyc database. These discoveries provided new insights for us to elucidate the potential functions of lung bacteriomes on growth and persistent state of MTB bacillus.
Interestingly, the MetaCyc pathway responsible for penicillin resistance was also enriched in TB-E samples (Supplemental Fig. 1), although penicillin was ineffective in anti-TB treatment. According to the MetaCyc database (metacyc.org), phyla Actinobacteria and Firmicutes are expected to possess the pathway responsible for penicillin resistance. Relative abundance of Actinobacteria and Firmicutes in TB-E were 22.0% and 9.2%, which were higher than those (Actinobacteria 17.8% and Firmicutes 8.4%) in TB-NE samples. This might be the main reason that the bacterial pathway responsible for penicillin resistance was relevant to TB-E samples. The mechanism causing penicillin resistance in bacteria is overexpression of low-affinity penicillin-binding proteins (PBPs) and β-lactamases [41]. The enrichment of this bacterial pathway may indicate that TB-E lesions are more likely to co-infect with other penicillin-resistant pathogens.
In TB-NE samples, bacterial MetaCyc pathway responsible for heme b synthesis was significantly enriched. Heme biosynthesis is generally present in the host, but the bacterial metabolic pathway HEME-BIOSYNTHESIS-II is also possessed in some bacterial members under aerobic condition according to the MetaCyc database. Heme is a vital iron-containing prosthetic group for hemoproteins in MTB bacillus to acquire iron nutrient from the host. Heme b biosynthesis is one of the metabolic pathways responsible for iron assimilation [42]. A previous study has reported that macrophages can ingest bacterial components [8], which might elevate the intracellular iron level of macrophages through absorbing bacterial irons. Elevated level of intracellular iron is a character of macrophage necrosis induced by MTB infection [43, 44]. Collectively, we thought lung bacteria, especially their ingested components in macrophages, might contribute to TB pathology through ferroptosis.
As for the remaining bacterial MetaCyc functions with statistically different enrichments between TB-E and TB-NE groups, their potential impacts on MTB bacillus or pulmonary TB disease have not been clearly reported yet, but they still provide useful foundation for future investigations.
Conclusion
This study innovatively demonstrates the characteristics of bacteriomes in lower respiratory tract using lung tissues. Bacterial composition and MetaCyc functions in TB lesions are strongly associated with the status of MTB excretion. Co-existing bacteria found in TB lesions can partially impact the survival of MTB and the development of TB pathology through metabolic functions. This study provide important bacterial information to develop novel strategies for prevention and treatment of pulmonary TB disease in the future.
Availability of data and materials
The 16S rDNA raw data were deposited in the National Center for Biotechnology Information (NCBI) at Sequence Read Archive (SRA) under BioProject accession number PRJNA747828.
Abbreviations
- TB:
-
Tuberculosis
- MTB:
-
Mycobacterium tuberculosis
- TB-E:
-
TB patients excreting bacteria
- TB-NE:
-
TB patients not excreting bacteria
- NT:
-
Normal lung tissues
- PICRUSt:
-
Phylogenetic investigation of communities by reconstruction of unobserved states
- ANOSIM:
-
Analysis of similarity
- WHO:
-
World Health Organization
- BMI:
-
Body mass index
- MAIT cells:
-
Mucosal-associated invariant T cells
- BAF:
-
Bronchoalveolar lavage fluid
- FFPE:
-
Formalin fixed and paraffin embedded
- AFB:
-
Acid Fast Bacilli
- HE:
-
Hematoxylin–eosin
- OTU:
-
Operational taxonomic unit
- PCoA:
-
Principal coordinates analysis
- ANOVO:
-
Analysis of variance
- PBP:
-
Penicillin-binding proteins
References
Telzak EE, Fazal BA, Pollard CL, Turett GS, Justman JE, Blum S. Factors influencing time to sputum conversion among patients with smear-positive pulmonary tuberculosis. Clin Infect Dis. 1997;25:666–70. https://doi.org/10.1111/j.1440-1843.2009.01598.x.
Falzon D, Schünemann HJ, Harausz E, González-Angulo L, Lienhardt C, Jaramillo E, Zignol M, Kasaeva T. World Health Organization treatment guidelines for drug-resistant tuberculosis. Eur Respir J. 2017;49:1602308. https://doi.org/10.1183/13993003.03300-2020.
Nguyen MVH, Levy NS, Ahuja SD, Trieu L, Proops DC, Achkar JM. Factors associated with sputum culture-negative vs culture-positive diagnosis of pulmonary tuberculosis. JAMA Netw Open. 2019;2:e187617–e187617. https://doi.org/10.1001/jamanetworkopen.2018.7617.
Sarathy JP, Dartois V. Caseum: a niche for Mycobacterium tuberculosis drug-tolerant persisters. Clin Microbiol Rev. 2020;33:e00159-e219. https://doi.org/10.1128/CMR.00159-19.
Cailleaux-Cezar M, Loredo C, Silva JRL, Conde MB. Impact of smoking on sputum culture conversion and pulmonary tuberculosis treatment outcomes in Brazil: a retrospective cohort study. J Bras Pneumol. 2018;44:99–105. https://doi.org/10.1590/s1806-37562017000000161.
Putri FA, Burhan E, Nawas A, Soepandi PZ, Sutoyo DK, Agustin H, Isbaniah F, Dowdy DW. Body mass index predictive of sputum culture conversion among MDR-TB patients in Indonesia. Int J Tuberc Lung D. 2014;18:564–70. https://doi.org/10.5588/ijtld.13.0602.
Dickson RP, Erb-Downward JR, Huffnagle GB. Towards an ecology of the lung: new conceptual models of pulmonary microbiology and pneumonia pathogenesis. Lancet Resp Med. 2014;2:238–46. https://doi.org/10.1016/S2213-2600(14)70028-1.
Nejman D, Livyatan I, Fuks G, Gavert N, Zwang Y, Geller LT, Rotter-Maskowitz A, Weiser R, Mallel G, et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science. 2020;368:973–80. https://doi.org/10.1126/science.aay9189.
Dumas A, Corral D, Colom A, Levillain F, Peixoto A, Hudrisier D, Poquet Y, Neyrolles O. The host microbiota contributes to early protection against lung colonization by Mycobacterium tuberculosis. Front Immunol. 2018;9:2656. https://doi.org/10.3389/fimmu.2018.02656.
Botero LE, Delgado-serrano L, Cepeda ML, Bustos JR, Anzola JM, Portillo PD, Robledo J, Zambrano MM. Respiratory tract clinical sample selection for microbiota analysis in patients with pulmonary tuberculosis. Microbiome. 2014;2:29–29. https://doi.org/10.1186/2049-2618-2-29.
Krishna P, Jain A, Bisen PS. Microbiome diversity in the sputum of patients with pulmonary tuberculosis. Eur J Clin Microbiol. 2016;35:1205–10. https://doi.org/10.1007/s10096-016-2654-4.
Ticlla MR, Hella J, Hiza H, Sasamalo M, Mhimbira F, Rutaihwa LK, Droz S, Schaller S, Reither K, Hilty M, et al. The sputum microbiome in pulmonary tuberculosis and its association with disease manifestations: a cross-sectional study. Front Microbiol. 2021;12: 633396. https://doi.org/10.3389/fmicb.2021.633396.
Hu YF, Cheng M, Liu B, Dong J, Sun LL, Yang J, Yang F, Chen XC, ** Q. Metagenomic analysis of the lung microbiome in pulmonary tuberculosis - a pilot study. Emerg Microbes Infec. 2020;9:1444–52. https://doi.org/10.1080/22221751.2020.1783188.
Schneeberger PH, Prescod J, Levy L, Hwang D, Martinu T, Coburn B. Microbiota analysis optimization for human bronchoalveolar lavage fluid. Microbiome. 2019;7:1–16. https://doi.org/10.1186/s40168-019-0755-x.
Caporason JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6. https://doi.org/10.1038/nmeth.f.303.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1. https://doi.org/10.1093/bioinformatics/btq461.
Navas-Molina JA, Peralta-Sánchez JM, González A, McMurdie PJ, Vázquez-Baeza Y, Xu Z, Ursell LK, Lauber C, Zhou H, Song SJ, et al. Advancing our understanding of the human microbiome using QIIME. Methods Enzymol. 2013;531:371–444. https://doi.org/10.1016/B978-0-12-407863-5.00019-8.
Douglas GM, Maffei VJ, Zaneveld JR, Yurgel SN, Brown JR, Taylor CM, Huttenhower C, Langille MG. PICRUSt2 for prediction of metagenome functions. Nat Biotechnol. 2020;38:685–8. https://doi.org/10.1038/s41587-020-0548-6.
Kahle DJ, Wickham H. ggmap: spatial visualization with ggplot2. R J. 2013;5:144.
Kolde R, Kolde MR. Package ‘pheatmap.’ R package. 2015;1:790.
Chen H, Boutros PC. VennDiagram: a package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinformatics. 2011;12:35.
Watts DJ, Strogatz SH. Collective dynamics of ‘small-world’ networks. Nature. 1998;393:440–2. https://doi.org/10.1038/30918.
Louca S, Polz MF, Mazel F, Albright MBN, Huber JA, O’Connor MI, Ackermann M, Hahn AS, Srivastava DS, Crowe SA, et al. Function and functional redundancy in microbial systems. Nat Ecol Evol. 2018;2:936–43. https://doi.org/10.1038/s41559-018-0519-1.
Cui ZL, Zhou YH, Li H, Zhang Y, Zhang SL, Tang SJ, Guo XH. Complex sputum microbial composition in patients with pulmonary tuberculosis. BMC Microbiol. 2012;12:276. https://doi.org/10.1186/1471-2180-12-276.
Dickson RP, Erb-Downward JR, Freeman CM, McCloskey L, Beck JM, Huffnagle B, Curtis JL. Spatial variation in the healthy human lung microbiome and the adapted island model of lung biogeography. Ann Am Thorac Soc. 2017;12:821–30. https://doi.org/10.1513/AnnalsATS.201501-029OC.
Baker JM, Hinkle KJ, McDonald RA, Brown CA, Falkowski NR, Huffnagle GB, Dickson RP. Whole lung tissue is the preferred sampling method for amplicon-based characterization of murine lung microbiota. Microbiome. 2021;9:1–14. https://doi.org/10.1186/s40168-021-01055-4.
Vázquez-Pérez JA, Carrillo CO, Iñiguez-García MA, Remero-Espinoza I, Márquez-García JE, Falcón LI, Torres M, Herrera MT. Alveolar microbiota profile in patients with human pulmonary tuberculosis and interstitial pneumonia. Microb Pathogenesis. 2020;139: 103851. https://doi.org/10.1016/j.micpath.2019.103851.
Lin PL, Flynn JL. The end of the binary era: revisiting the spectrum of tuberculosis. J Immunol. 2018;201:2541–8. https://doi.org/10.4049/jimmunol.1800993.
Zhou Y, Lin F, Cui Z, Zhang X, Hu C, Shen T, Chen C, Zhang X, Guo X. Correlation between either cupriavidus or porphyromonas and primary pulmonary tuberculosis found by analysing the microbiota in patients’ bronchoalveolar lavage fluid. PLoS ONE. 2015;10: e0124194. https://doi.org/10.1371/journal.pone.0124194.
Hong BY, Maulén NP, Adami AJ, Granados H, Balcells ME, Cervantes J. Microbiome changes during tuberculosis and antituberculous therapy. Clin Microbiol Rev. 2016;29:915–26. https://doi.org/10.1128/CMR.00096-15.
Sloan WT, Lunn M, Woodcock S, Head IM, Nee S, Curtis TP. Quantifying the roles of immigration and chance in sha** prokaryote community structure. Environ Microbiol. 2006;8:732–40. https://doi.org/10.1111/j.1462-2920.2005.00956.x.
Ofiteru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, Sloan WT. Combined niche and neutral effects in a microbial wastewater treatment community. PNAS. 2010;107:15345–50. https://doi.org/10.1073/pnas.1000604107.
Fernandez-Gonzalez N, Huber JA, Vallino JJ. Microbial communities are well adapted to disturbances in energy input. mSystems. 2016;1:e00117-16. https://doi.org/10.1128/mSystems.00117-16.
Ponomarova O, Patil KR. Metabolic interactions in microbial communities: untangling the Gordian knot. Curr Opin Microbiol. 2015;27:37–44. https://doi.org/10.1016/j.mib.2015.06.014.
Girvan MS, Campbell CD, Killham K, Prosser JI, Glover LA. Bacterial diversity promotes community stability and functional resilience after perturbation. Environ Microbiol. 2005;7:301–13. https://doi.org/10.1111/j.1462-2920.2005.00695.x.
Peter H, Beier S, Bertilsson S, Lindström ES, Langenheder S, Tranvik LJ. Function-specific response to depletion of microbial diversity. ISME J. 2011;5:351–61. https://doi.org/10.1038/ismej.2010.119.
Bashiri G, Nigon LV, Jirgis ENM, Ho NAT, Stanborough T, Dawes SS, Baker EN, Bulloch EMM, Johnston JM. Allosteric regulation of menaquinone (vitamin K2) biosynthesis in the human pathogen Mycobacterium tuberculosis. J Biol Chem. 2020;295:3759–70. https://doi.org/10.1074/jbc.RA119.012158.
Debnath J, Siricilla S, Wan B, Crick DC, Lenaerts AJ, Franzblau SG, Kurosu M. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J Med Chem. 2012;55:3739–55. https://doi.org/10.1021/jm201608g.
Coggins JR, Abell C, Evans LB, Frederickson M, Robinson DA, Roszak AW, Lapthorn AP. Experiences with the shikimate-pathway enzymes as targets for rational drug design. Biochem Soc Trans. 2003;31:548–51. https://doi.org/10.1042/bst0310548.
Khanapur M, Alvala M, Prabhakar M, Kumar KS, Edwin RK, Sir Saranya PSVK, Patel RK, Bulusu G, Misra P, Pal M. Mycobacterium tuberculosis chorismate mutase: a potential target for TB. Bioorganic Med Chem. 2017;25:1725–36. https://doi.org/10.1016/j.bmc.2017.02.001.
Massova I, Mobashery S. Kinship and diversification of bacterial penicillin-binding proteins and β-lactamases. Antimicrob Agents Ch. 1998;42:1–17. https://doi.org/10.1128/AAC.42.1.1.
Kurthkoti K, Amin H, Marakalala MJ, Ghanny S, Subbian S, Sakatos A, Livny J, Fortune SM, Berney M, Rodriguez GM. The capacity of Mycobacterium tuberculosis to survive iron starvation might enable it to persist in iron-deprived microenvironments of human granulomas. mBio. 2017;8:e01092-17. https://doi.org/10.1128/mBio.01092-17.
Amaral EP, Costa DL, Namasivayam S, Riteau N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB, Sher A. A major role for ferroptosis in Mycobacterium tuberculosisinduced cell death and tissue necrosis. J Exp Med. 2019;216:556–70. https://doi.org/10.1084/jem.20181776.
Meunier E, Neyrolles O. Die another way: ferroptosis drives tuberculosis pathology. J Exp Med. 2019;216:471–3. https://doi.org/10.1084/jem.20190038.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Grant Number: 31900098, 82072381), Bei**g Municipal Administration of Hospitals Incubating Program (code: PX2020066), Bei**g Municipal Science and Technology Project (Z181100001918027, Z191100006619079), and Tongzhou High-level Technique Talents Program (YHLD2018006).
Author information
Authors and Affiliations
Contributions
W.D. conceived the project, analyzed the original high-throughput sequencing date, interpreted the data and wrote the manuscript. N.C. conceived the project and interpreted the data. Y.Z., L.Z., J.C., Z.L. and K.L. conducted part of the lab work. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the ethical and institutional review boards for human investigation at Bei**g Chest Hospital, Capital Medical University (Number: 2020-keyan-linjian15). All methods in this study were performed in accordance with the relevant guideline and regulations. Written informed consent was provided by all the participants or their guardians for participation in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Supplemental Table 1. Values of bacterial MetaCyc pathways of all TB lesions (40 TB-E and 42 TB-NE).
Additional file 2:
Supplemental Table 2. Bacterial metabolic pathways that are significantly down/up regulated in TB-E compared to TB-NE.
Additional file 3:
Supplemental Figure 1. Boxplots showing the abundance of Menaquinone biosynthesis, Chorismate biosynthesis, Penicillin resistance and Heme biosynthesis in TB-E and TB-NE samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, W., Zhao, Y., Zhang, L. et al. Bacteriomes in lesions of pulmonary tuberculosis and its association with status of Mycobacterium tuberculosis excretion. BMC Microbiol 22, 280 (2022). https://doi.org/10.1186/s12866-022-02698-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-022-02698-5