
Abstract
Background
Specific pathogen-free ducks are a valuable laboratory resource for waterfowl disease research and poultry vaccine development. High throughput sequencing allows the systematic identification of structural variants in genomes. Copy number variation (CNV) can explain the variation of important duck genetic traits. Herein, the genome-wide CNVs of the three experimental duck species in China (**ding ducks (JD), Shaoxing ducks (SX), and Fujian Shanma ducks (SM)) were characterized using resequencing to determine their genetic characteristics and selection signatures.
Results
We obtained 4,810 CNV regions (CNVRs) by merging 73,012 CNVs, covering 4.2% of the duck genome. Functional analysis revealed that the shared CNVR-harbored genes were significantly enriched for 31 gene ontology terms and 16 Kyoto Encyclopedia of Genes and Genomes pathways (e.g., olfactory transduction and immune system). Based on the genome-wide fixation index for each CNVR, growth (SPAG17 and PTH1R), disease resistance (CATHL3 and DMBT1), and thermoregulation (TRPC4 and SLIT3) candidate genes were identified in strongly selected signatures specific to JD, SM, and SX, respectively.
Conclusions
In conclusion, we investigated the genome-wide distribution of experimental duck CNVs, providing a reference to establish the genetic basis of different phenotypic traits, thus contributing to the management of experimental animal genetic resources.
Similar content being viewed by others


Background
Genetic variation is an important genetic basis for individual differences. Copy number variation (CNV) is a type of genetic variation that varies from 50 kb to a few Mb in size compared with the reference genome sequence of an organism, particularly a deletion event or duplication type affecting many base pairs [1]. Neighboring CNV areas with overlap** regions can be merged into one large genomic segment referred as a copy number variant region (CNVR). CNV is a complementary genetic variant to a single nucleotide polymorphism (SNP), which has a more substantial impact on gene expression and function. For example, changing the genetic structure and dose, destroying a coding sequence, interfering with long-term gene regulation, and exposure of recessive genes, all of which have important implications for animal phenotypic polymorphism, disease susceptibility, and evolutionary adaptation [2,3,4].
With the development of large-scale human CNV research, substantial progress has been made in CNV detection in livestock and poultry species, including cattle (Bos taurus) [5, 6], pigs (Sus scrofa) [7], goats (Capra hircus) [10, 11], meat quality [12], reproduction [13], immune response [14], disease [15], and environmental adaptations [16]. Moreover, CNVs provide resources towards the creation of new genes [17]. Compared with SNP chips and array comparative genomic hybridization (aCGH) microarrays chips, whole genome resequencing (WGRS) technology is more comprehensive and accurate for genome level recognition of CNVs, thereby improving the accuracy of functional genetic prediction [18].
Duck (Anas platyrhynchos) is the most widespread and agriculturally important waterfowl species in the world, providing significant economic benefits from its use as a high-quality source of meat, eggs, and feathers [19]. Furthermore, ducks are hosts for most avian diseases and have been shown to undergo high morbidity, long-distance transmission and carry multiple viruses [20, 21]. Consequently, Specific Pathogen Free (SPF) ducks, which have been artificially bred to carry controlled microorganisms and have a clear genetic background, are important experimental materials for avian pathology research and the production of avian-derived biological products [22, 23]. Characterization of whole-genome sequence variation in SPF ducks and the identification of phenotype-related functional variants are crucial to assess their genetic quality and are necessary to guide future genome-assisted breeding and disease studies.
With the availability of duck reference genome sequences, studies have successfully applied SNP loci identified by WGRS technology in population structure analysis [24], trait localization [25], and population evolution [19] of ducks. However, CNVs in the duck genome have not been thoroughly studied on a genome wide basis. After Skinner et al. [26] obtained the first genomic CNV map of ducks using the aCGH detection method, only Xu et al. [27] has explored the CNVs associated with the number of cervical vertebrae in Pekin ducks using genome-wide association analysis. Herein, the CNVs of three representative experimental ducks, **ding duck (JD), Shaoxing duck (SX), and Fujian Shanma duck (SM), were analyzed using the WGRS technique for the first time. They are often used as ancestral generations to breed new varieties because of their excellent fecundity and adaptive performance. Besides, the successful breeding of JD, SX and SM populations has led to their wide use in research related to avian pathogens, such as Newcastle disease virus [28], avian influenza virus [29], and avian reovirus [30]. Our main objectives were to characterize the genome wide CNV variation within and among populations, and identify CNVs and related functional genes associated with different phenotypic traits in each experimental duck. A significant number of experimental duck CNVs and candidate CNVRs were identified, which will provide a valuable resource for the genetic characterization of different experimental duck populations.
Materials and methods
Sample collection
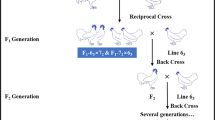
SPF experimental ducks in this study represent the current breeding ducks in China and were maintained in an isolation environment at the National Poultry Laboratory Animal Resource Center (LARC). The cultivation of SPF ducks requires a series of strict processes (Fig. 1). In brief, all ducks were raised based on high-quality breeding eggs (SX are introduced from Shaoxing shelduck ancestor duck eggs of the Academy of Agricultural Sciences in Zhejiang Province; JD are introduced from the National Waterfowl Base Resource Library in Taizhou, Jiangsu; SM are introduced from the Shelduck Original Breeding Farm in Longyan City, Fujian), reared in positive pressure isolators in a barrier environment. The detection of pathogenic microorganisms was carried out in regular population surveys to eliminate positive individuals and ducks were confirmed to be free of duck hepatitis virus I, duck plague virus, duck circovirus, duck tembusu virus, goose parvovirus, avian leukosis viruses, avian reovirus, avian influenza virus (H5 subtype (Re8 strain), H7 subtype, H9 subtype), Newcastle disease virus, and avian adenovirus II. Using the wing vein method, we collected blood samples from 30 SPF ducks of similar body weight at 42 weeks old, including JD (5♀, 5♂), SX (5♀, 5♂), and SM (5♀, 5♂). Samples were immediately snap-frozen in liquid nitrogen for further extraction of genomic DNA. Table S1 shows the phenotypic descriptions of three breeds.

Cultivation flow chart of SPF ducks
Sequencing data processing
Genomic DNA was extracted from blood samples and paired-end libraries were constructed (insert size of 300–400 bp) using the Illumina NovaSeq 6000 platform (San Diego, CA, USA). Quality control was used to filter the reads to remove adapters and low-quality reads. The filtered raw reads were further processed to obtain high quality clean reads based on three strict filtering criteria: (1) removing reads with ≥ 10% unidentified nucleotides; (2) removing reads with > 50% bases having phred quality scores of ≤ 20; and (3) removing reads aligned to the barcode adapter. The clean reads were mapped to the Anas platyrhynchos reference genome obtained from the NCBI (https://www.ncbi.nlm.nih.gov/assembly/GCF_015476345.1) using the BWA software, with the following parameters: mem -t 4 -k 32 -M [31]. Duplicates were removed using SAMtools software [32]. If multiple read pairs had identical external coordinates, only the pair with the highest map** quality were retained.
Detection of CNVs and CNVRs
We firstly used CNVnator software to detect the CNVs in each duck genome sample [33]. Quality control was performed on the raw CNVs of each sample. The screening criteria included a p-value < 0.01 (e-val1 calculated using t-test statistics), size > 1 kb. CNV_type was judged based on the read depth (RD) value (deletion: RD < 0.7; duplication: RD > 1.3). Then, CNVcaller was used to detect population-level CNVRs [34]. To obtain high-confidence CNVs and CNVRs, we performed the following quality control procedures: (1) The CNVs of 10 samples from each group were fused using the “Merge” command of BEDTools [35]. (2) When overlap** sequences were at least > 1 bp along their genomic coordinates, we used the “intersect” command in BEDTools to merge multiple adjacent CNVs between individuals within a population into one CNVR and discarded each population or CNVRs that contained only one CNV in the metapopulation. We defined CNVRs containing only deletions as deleted CNVRs, duplicated CNVRs as duplicated CNVRs, and CNVRs containing both deletions and duplicates as complex CNVRs.
Functional enrichment analysis of CNVR-harboring genes
CNVR-harboring genes were searched in the Anas platyrhynchos reference genome, and completely and partially (≥ 50%) overlap** genes were retained for subsequent analysis. Functional enrichment analysis using gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) [36], performed using the online tool DAVID (https://david.ncifcrf.gov/). A false discovery rate (FDR) < 0.05 was considered to indicate significant enrichment of candidate genes.
Population genetics of CNVRs
Levels of genetic differentiation among populations were evaluated by using the Fixation index (FST) method [37], using the -weir-fst-pop option in VCFtools [38]. Functional enrichment analysis was performed on the top 5% of CNVR loci showing extremely high FST values and tested whether these “outlier” loci were associated with important traits in ducks.
Results
Sequencing and CNV detection
Using Illumina paired-end sequencing technology, we obtained high-quality next generation sequencing data for 30 experimental ducks (Additional file 2: Table S2). The mapped read depth ranged from 19.65× to 28.33×, with an average depth of 22.65× per sample, indicating that these data were sufficient for further analysis (Additional file 3: Table S3). We detected a total of 73,012 CNVs, including 26,432 “duplication” events and 46,580 “deletion” events. The sizes of all the CNVs showed an L-shaped distribution (median size = 7.8 kb, average size = 19.0 kb) (Fig. 2a and Additional file 4: Table S4). At the individual level, we found an average of 1956 CNVs per duck genome, ranging from 1707 to 2398 (Fig. 2b and Additional file 5: Table S5). By merging overlap** CNVs, a total of 4,810 CNVRs were obtained, covering 4.2% of the duck genome (Additional file 6: Table S6). Among them, 2,263; 2,127; and 2,128 CNVRs were obtained in the SM, JD and SX ducks, respectively (Additional file 7: Fig. S1). There was a significant positive linear relationship between the number of CNVRs and the corresponding autosomal size (R2 = 0.85, Fig. 3).

Genome-wide characterization of CNVs in the duck genome. (a) A histogram of the distribution of CNV length. (b) Total length and the total amount of CNVs identified in each sample

Genomic landscape of CNVRs. (a) A map of CNVRs in the duck genome; Two types of CNVR were identified, including duplication (yellow) and deletion (green). (b) Correlation between CNVR counts and chromosome length
Functional annotation of the identified CNVRs
From the genome annotation, there were 750, 97, and 92 CNVR-harboring genes detected only in SM, JD, and SX, respectively, while 2675 CNVR-harboring genes were detected in all three populations (Additional file 8: Fig. S2). The functional enrichment analysis showed that 31 GO terms were enriched in the CNVRharboring genes shared by the three populations, comprising 6 biological processes, 9 cellular components, and 16 molecular functions. These GO terms were mainly related to olfactory receptor activity (GO:0004984) and signaling receptor activity (GO:0004888, GO:0060089, GO:0038023, and GO:0004930) (Additional file 9: Table S7). The KEGG pathway analysis identified 16 significantly enriched pathways, including olfactory transduction (ko04740) and the immune system (ko05320, ko04612, and ko04650) (Fig. 4 and Additional file 10: Table S8). Furthermore, we performed functional enrichment analysis of specific CNVR-harboring genes in the three duck populations. In particular, the CNVR-harboring genes specifically distributed in SM were mainly involved in oxygen transporter activity (GO:0005344) and oxygen binding (GO:0019825).

KEGG pathway enrichment analysis (www.kegg.jp/kegg/kegg1.html). (a) Top 20 enriched signaling pathways of CNVR-harbored genes shared among the three duck populations. (b) Top 20 enriched signaling pathways for JD-, (c) SM-, and (d) SX-specific CNVR-harbored genes
Population genetics of CNVRs
Through estimating the genome-wide FST to detect the CNVRs that were genetically differentiated in each duck population, we were able to perform the following comparisons: CNVRs from JD compared with those from SX and SM, CNVRs from SM compared with those from JD and SX, and CNVRs from SX compared with those from JD and SM. Based on the top 5% of the FST distribution, 159 outlier loci that overlapped with 56 genes were considered highly divergent in JD (Fig. 5a and Additional file 11: Table S9). The functional analysis identified three significantly enriched pathways, including the C-type lectin receptor signaling pathway (ko04625), tuberculosis (ko05152) and endocrine and other factor-regulated calcium reabsorption (ko04961) (Fig. 5b and Additional file 12: Table S10). Among all CNVRs analyzed, we identified a 2,000 bp deletion (chromosome 1: 83,517,601–83,519,600 bp) in SPAG17 (encoding sperm-associated antigen 17) and a 1,600 bp deletion (chromosome 2: 163,037,601–163,039,200 bp) in PTH1R (encoding parathyroid hormone 1 receptor). The frequencies of their deletion were high in JD, but lower in the other populations (Fig. 5c).

Comparative genomic analysis for JD vs. SX and SM using population fixation index (FST). (a) Manhattan plot of genome-wide FST on each CNVR locus between for JD vs. SX and SM. (b) Top twenty enriched KEGG pathways for the genes overlapped with highly differentiated CNVRs for JD vs. SX and SM (www.kegg.jp/kegg/kegg1.html). (c) Allele frequencies of SPAG17 and PTH1R
Based on the top 5% of the empirical FST distribution, 159 outlier loci that overlapped with 51 genes were considered highly divergent in SM (Fig. 6a and Additional file 11: Table S9). KEGG pathway analysis showed that the enriched differentiated CNVR genes were mainly associated with viral myocarditis (ko05416), Staphylococcus aureus infection (ko05150) and protein digestion and absorption (ko04974) (Fig. 6b and Additional file 12: Table S10). The genome-wide distribution of FST showed that the most significantly variation was a 3,200 bp deletion (chromosome 2: 163,312,001–163,315,200 bp) that overlapped with the CATHL3 gene (encoding cathelicidin 3) and a 3,600 bp duplication (chromosome 5: 55,372,001–55,375,600 bp) overlap** with the DMBT1 gene (encoding deleted in malignant brain tumors 1). The frequencies of these CNVRs in SM were lower relative to that those in the other populations (Fig. 6c).

Comparative genomic analysis for SM vs. JD and SX using population fixation index (FST). (a) Manhattan plot of genome-wide FST on each CNVR locus between for SM vs. JD and SX. (b) Top twenty enriched KEGG pathways for the genes overlapped with highly differentiated CNVRs for SM vs. JD and SX (www.kegg.jp/kegg/kegg1.html). (c) Allele frequencies of CATHL3 and DMBT1
In the selective sweep analyses, we identified 161 outlier loci overlap** with 65 genes that were considered highly divergent in SX (Fig. 7a and Additional file 11: Table S9). Functional analysis revealed that genes overlap** with differentiated CNVRs were enriched in autoimmune thyroid disease (ko05320) and glycerophospholipid metabolism (ko00564) (Fig. 7b and Additional file 12: Table S10). Additionally, we identified the strongest selection signal as a 2,000 bp deletion (chromosome 14: 15,796,801–15,798,800 bp) containing the SLIT3 gene (encoding slit guidance ligand 3) and a 2,400 bp deletion (chromosome 1: 181,252,801–181,255,200 bp) containing the TRPC4 gene (encoding transient receptor potential channel 4), the deletion frequencies of which were high in SX, but much lower in the other populations (Fig. 7c).

Comparative genomic analysis for SX vs. JD and SM using population fixation index (FST). (a) Manhattan plot of genome-wide FST on each CNVR locus between for SX vs. JD and SM. (b) Top twenty enriched KEGG pathways for the genes overlapped with highly differentiated CNVRs for SX vs. JD and SM (www.kegg.jp/kegg/kegg1.html). (c) Allele frequencies of TRPC4 and SLIT3
Discussion
Understanding the genetic basis of phenotypic differences is a major theme in animal science. CNVs, as important sources of genetic diversity, have attracted widespread attention in the last decade because of their dramatic phenotypic consequences [39, 40]. Studies have shown that the average size of avian genomes and the range of variation in genome size are the smallest among all vertebrate groups (http://www.genomesize.com) and that the overall karyotype structure is highly conserved [41]. In addition, the number of CNVs is also lower in avian genomes compared with that in mammalian genomes [42]. Therefore, avian genomes are particularly suitable to analyze CNVs because of their unique combination of features [41]. However, research related to avian CNVs (especially in ducks) is scarce. Herein, we detected the CNVs of three experimental duck species in China based on the WGRS technique, which identified 2,263; 2,127; and 2,128 CNVRs for SM, JD, and SX ducks respectively by merging 73,012 CNVs from all duck samples. Compared with previous studies based on 600 K SNP chip array chicken CNV profiling [43], we detected about twice as many CNVRs per duck population, which is consistent with comparative genomics studies of chickens and Peking ducks reported by a previous study [26]. Furthermore, we found that the CNVRs accounted for 4.2% of the duck reference genome, whereas they accounted for 5.12% in chickens [43], 6.2% in yaks (Bos grunniens) [55] and Tibetan chickens [56] to explain their adaptation to hypoxia. This coincides with the actual situation of this duck population. The production area of SM in Longyan Reserve in Fujian Province is mostly composed of mountainous areas, suggesting that SM can survive well in an environment with a limited oxygen supply.
Selection signature analysis based on sequencing data can reveal genomic regions that have undergone artificial selection and environmental change during local adaptation and evolution [57]. To screen for selection regions and genes specific to each population, the FST values for one population of experimental ducks compared with those of the other populations. Herein, the CNVR harboring genes SPAG17 and PTH1R showed significant differentiation in JD. SPAG17 encodes a multifunctional cytoplasmic protein that not only affects reproduction, but also is used extensively in the analysis of body-measurement traits related to skeletal development [58]. SPAG17 plays a crucial role in determining human body height. SNPs of SPAG17 have been reported to be associated with height and idiopathic short stature in infants [59], children [60], and adults [61]. In the livestock industry, SPAG17 expression is often used as growth trait data to guide the scientific raising and breeding of animals, as reported in goats [58] and cattle [62]. PTH1R also plays an important role in skeletal homeostasis. After PTH activation of PTH1R, it mediates catabolic and anabolic processes in bone. PTHR1 gene mutation causes Jansen’s metaphyseal chondrodysplasia [63]. In another study, a 51 bp indel polymorphism in the PTH1R gene was associated with growth and carcass traits in chickens [64]. Therefore, the identification SPAG17 and PTH1R further deepened our understanding of the genetic mechanisms underlying growth traits in JD.
Although related issues have been extensively studied through SNPs, there have been few reports on CNVR-based selection signals for adaptation to disease resistance in humans and animals [65, 66]. In the present study, we highlighted genes (CATHL3 and DMBT) that overlapped highly differentiated CNVRs between SM and other duck populations. CATHL3 is a small cationic antimicrobial peptide with effective activity against a wide range of pathogens, including bacteria, viruses, and fungi [67]. Previous studies have confirmed that CATHL3 is a potential candidate gene related to disease resistance studies in humans [68] and Gir cattle (Bos indicus) [69]. Similarly, DMBT1, a member of the scavenger receptor cysteine-rich super family, is considered to play a role in tumorigenesis and pathogen defense [70]. A DMBT1-harbored SNP selection signal provides evidence of a bovine tuberculosis (bTB) susceptibility gene in cattle breeds [65]. SM inhabits a mountainous area that has been relatively closed to transportation for a long time, acting as a natural barrier to some extent. In addition, SM exists mainly in free-ranging populations with less vaccination during the breeding process. Therefore, we speculated that CNVR-harbored CATHL3 and DMBT1 have undergone natural selection by mountain ecology in SM, with possible importance in disease resistance. Further studies are warranted to characterize the causal relationship between these genes and disease resistance in SM.
Temperature stress (high or low temperatures) is one of the most serious environmental challenges facing poultry worldwide, with negative effects on duck health, welfare and productivity. Organisms can assess changes in environmental temperature to produce certain physiological and behavioral responses that benefit survival. The activation of certain ion channels of the transient receptor potential (TRP) family by changes in ambient temperature, as well as the identification of their heterogeneous expression patterns and heterogeneous temperature sensitivity, have triggered the interest of researchers to evaluate these proteins as candidate endogenous thermosensors [71]. TRPC4 has been identified as a promising molecular target for body temperature management. Loss-of-function studies of TRPC4 demonstrated its function in GABAergic warm sensitive neurons, resulting in extra deficits in basal temperature setting, warm defense, and fever responses [72]. Recent studies have reported that TRPC4 is associated with thermoregulation [73] in cattle and cold adaptation [74] in Arctic dogs. In addition, secretion of the macrophage cytokine SLIT3 by adipose tissue macrophages enhances cold adaptation via stimulating sympathetic nerves and thermogenesis in mice (Mus musculus) [75]. In this study, the highly differentiated CNVRs between SX and the other duck populations overlapped with TRPC4 and SLIT3. We hypothesized that TRPC4 and SLIT3 might be involved in thermoregulation in SX. Furthermore, functional analysis revealed that the autoimmune thyroid disease pathway was the most significantly enriched among all pathways for SX-differentiated CNVR genes. Studies have shown that thyroid disorders potentially interfere with the normal regulation of body temperature in humans [76]. Thyroid hormone synthesis is increased in birds and mammals in cold environments. The size and activity of the thyroid also increase especially at low temperatures [74, 77]. It has been reported that the expression of the TPO gene (encoding thyroid peroxidase, a key factor of the autoimmune thyroid disease pathway) is up-regulated in Bashang long-tail chicken (BS) and Rhode Island red chickens (RIR) in cold environments [78]. Therefore, we speculated that this pathway might also be related to the thermoregulation of SX. However, more functional experiments are necessary to fully reveal their biological functions.
Conclusion
In the present study, the first resequencing based CNV map of experimental SPF ducks was developed. Functional enrichment analysis of the identified genes in shared CNVRs revealed several underlying biological processes responsible for olfactory receptors and the immune system of experimental ducks. Selective sweep analysis showed that growth (SPAG17 and PTH1R), disease resistance (CATHL3 and DMBT1), and thermoregulation (TRPC4 and SLIT3) candidate gene were identified in strongly selected signatures specific to JD, SM, and SX, respectively. Although these phenotype-associated genes need to be further validated by biological experiments, our findings provide valuable information to identify the molecular basis of important phenotypic variations in experimental ducks.
Data Availability
The raw sequence data files from this study have been deposited in SRA and BioProject ID is PRJNA896757 (https://www.ncbi.nlm.nih.gov/sra/PRJNA896757).
Abbreviations
- CNV:
-
Copy number variation
- CNVRs:
-
Copy number variation regions
- JD:
-
**ding ducks
- SX:
-
Shaoxing ducks
- SM:
-
Fujian Shanma ducks
- SPF:
-
Specific pathogen-free
- SNP:
-
Single nucleotide polymorphism
- WGRS:
-
Whole-genome resequencing
- aCGH:
-
Array comparative genomic hybridization
- RD:
-
Read depth
- F ST :
-
Fixation index
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- SPAG17 :
-
Sperm-associated antigen 17
- PTH1R :
-
Parathyroid hormone 1 receptor
- CATHL3 :
-
Cathelicidin 3
- DMBT1 :
-
Deleted in malignant brain tumors 1
- SLIT3 :
-
Slit guidance ligand 3
- TRPC4 :
-
Transient receptor potential channel 4
References
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, et al. Map** copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65.
Zhang RQ, Wang JJ, Zhang T, Zhai HL, Shen W. Copy-number variation in goat genome sequence: a comparative analysis of the different litter size trait groups. Gene. 2019;696:40–6.
Chain FJ, Feulner PG, Panchal M, Eizaguirre C, Samonte IE, Kalbe M, et al. Extensive copy-number variation of young genes across stickleback populations. PLoS Genet. 2014;10(12):e1004830.
Zhang Z, Zhong H, Lin S, Liang L, Ye S, Xu Z, et al. Polymorphisms of AMY1A gene and their association with growth, carcass traits and feed intake efficiency in chickens. Genomics. 2021;113(2):583–94.
Mei C, Junjvlieke Z, Raza S, Wang H, Cheng G, Zhao C, et al. Copy number variation detection in Chinese indigenous cattle by whole genome sequencing. Genomics. 2020;112(1):831–6.
Liu GE, Bickhart DM. Copy number variation in the cattle genome. Funct Integr Genomics. 2012;12(4):609–24.
Ding R, Zhuang Z, Qiu Y, Wang X, Wu J, Zhou S, et al. A composite strategy of genome-wide association study and copy number variation analysis for carcass traits in a Duroc pig population. BMC Genomics. 2022;23(1):590.
Dong Y, Zhang X, **e M, Arefnezhad B, Wang Z, Wang W, et al. Reference genome of wild goat (capra aegagrus) and sequencing of goat breeds provide insight into genic basis of goat domestication. BMC Genomics. 2015;16(1):431.
Li X, Yang J, Shen M, **e XL, Liu GJ, Xu YX, et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat Commun. 2020;11(1):2815.
Weich K, Affolter V, York D, Rebhun R, Grahn R, Kallenberg A, et al. Pigment intensity in dogs is associated with a copy number variant upstream of KITLG. Genes (Basel). 2020;11(1):75.
Han JL, Yang M, Yue YJ, Guo TT, Liu JB, Niu CE, et al. Analysis of agouti signaling protein (ASIP) gene polymorphisms and association with coat color in tibetan sheep (Ovis aries). Genet Mol Res. 2015;14(1):1200–9.
Stothard P, Choi JW, Basu U, Sumner-Thomson JM, Meng Y, Liao X, et al. Whole genome resequencing of black Angus and Holstein cattle for SNP and CNV discovery. BMC Genomics. 2011;12:559.
Ahmad SF, Singh A, Panda S, Malla WA, Kumar A, Dutt T. Genome-wide elucidation of CNV regions and their association with production and reproduction traits in composite Vrindavani cattle. Gene. 2022;830:146510.
Liu GE, Brown T, Hebert DA, Cardone MF, Hou Y, Choudhary RK, et al. Initial analysis of copy number variations in cattle selected for resistance or susceptibility to intestinal nematodes. Mamm Genome. 2011;22(1–2):111–21.
Luo J, Yu Y, Mitra A, Chang S, Zhang H, Liu G, et al. Genome-wide copy number variant analysis in inbred chickens lines with different susceptibility to Marek’s Disease. (Bethesda). 2013;G3(2):217–23.
Buggiotti L, Yudin NS, Larkin DM. Copy number variants in two northernmost cattle breeds are related to their adaptive phenotypes. Genes (Basel). 2022;13(9):1595.
Shwan N, Louzada S, Yang F, Armour J. Recurrent rearrangements of human amylase genes create multiple Independent CNV series. Hum Mutat. 2017;38(5):532–9.
Tam V, Patel N, Turcotte M, Bossé Y, Paré G, Meyre D. Benefits and limitations of genome-wide association studies. Nat Rev Genet. 2019;20(8):467–84.
Zhang Z, Jia Y, Almeida P, Mank JE, van Tuinen M, Wang Q, et al. Whole-genome resequencing reveals signatures of selection and timing of duck domestication. GigaScience. 2018;7(4):giy027.
Zhao L, Niu Y, Lu T, Yin H, Zhang Y, Xu L, et al. Metagenomic analysis of the **ding duck fecal virome. Curr Microbiol. 2018;75(6):658–65.
Chen GQ, Zhuang QY, Wang KC, Liu S, Shao JZ, Jiang WM, et al. Identification and survey of a novel avian coronavirus in ducks. PLoS ONE. 2013;8(8):e72918.
Wu F, Lu F, Fan X, Pan Q, Zhao S, Sun H, et al. Development of a live attenuated duck hepatitis a virus type 3 vaccine (strain SD70). Vaccine. 2020;38(30):4695–703.
Liu X, Kong X. Isolation, identification and attenuation of a pathogenic duck Hepatitis virus type 1 in China, and complete genomic sequence comparison between the embryo-passaged, attenuated derivatives and their parent. Pol J Vet Sci. 2019;22(1):163–71.
Feng P, Zeng T, Yang H, Chen G, Du J, Chen L, et al. Whole-genome resequencing provides insights into the population structure and domestication signatures of ducks in eastern China. BMC Genomics. 2021;22(1):401.
Zhou Z, Li M, Cheng H, Fan W, Yuan Z, Gao Q, et al. An intercross population study reveals genes associated with body size and plumage color in ducks. Nat Commun. 2018;9(1):2648.
Skinner BM, Robertson LB, Tempest HG, Langley EJ, Ioannou D, Fowler KE, et al. Comparative genomics in chicken and Pekin duck using FISH map** and microarray analysis. BMC Genomics. 2009;10:357.
Xu Y, Hu J, Fan W, Liu H, Zhang Y, Guo Z, et al. Genome-wide association analysis reveals 6 copy number variations associated with the number of cervical vertebrae in Pekin ducks. Front Cell Dev Biol. 2022;10:1041088.
Dai Y, Cheng X, Liu M, Shen X, Li J, Yu S, et al. Experimental Infection of duck origin virulent Newcastle Disease virus strain in ducks. BMC Vet Res. 2014;10:164.
Chai H, Wang Y, Hua Y, Guan X, Li Y, Liu J. Evaluation of specific pathogen-free ducks infected with the highly pathogenic avian Influenza virus h5n1 subtype derived from wild birds. Pak J Zool. 2014;46:625–31.
Yu K, Ti J, Lu X, Pan L, Liu L, Gao Y, et al. Novel duck reovirus exhibits pathogenicity to specific pathogen-free chickens by the subcutaneous route. Sci Rep. 2021;11(1):11769.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–9.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Wang X, Zheng Z, Cai Y, Chen T, Li C, Fu W, et al. CNVcaller: highly efficient and widely applicable software for detecting copy number variations in large populations. GigaScience. 2017;6(12):1–12.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38(6):1358–70.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456(7218):18–21.
Clop A, Vidal O, Amills M. Copy number variation in the genomes of domestic animals. Anim Genet. 2012;43(5):503–17.
Völker M, Backström N, Skinner BM, Langley EJ, Bunzey SK, Ellegren H, et al. Copy number variation, chromosome rearrangement, and their association with recombination during avian evolution. Genome Res. 2010;20(4):503–11.
Griffin DK, Robertson LB, Tempest HG, Vignal A, Fillon V, Crooijmans RP, et al. Whole genome comparative studies between chicken and Turkey and their implications for avian genome evolution. BMC Genomics. 2008;9:168.
Gorla E, Cozzi MC, Román-Ponce SI, Ruiz López FJ, Vega-Murillo VE, Cerolini S, et al. Genomic variability in Mexican chicken population using copy number variants. BMC Genet. 2017;18(1):61.
Wang H, Chai Z, Hu D, Ji Q, **n J, Zhang C, et al. A global analysis of CNVs in diverse yak populations using whole-genome resequencing. BMC Genomics. 2019;20(1):61.
Guo J, Zhong J, Liu GE, Yang L, Li L, Chen G, et al. Identification and population genetic analyses of copy number variations in six domestic goat breeds and Bezoar ibexes using next-generation sequencing. BMC Genomics. 2020;21(1):840.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9.
Spehr M, Munger SD. Olfactory receptors: G protein-coupled receptors and beyond. J Neurochem. 2009;109(6):1570–83.
Abo-Ismail MK, Vander Voort G, Squires JJ, Swanson KC, Mandell IB, Liao X, et al. Single nucleotide polymorphisms for feed efficiency and performance in crossbred beef cattle. BMC Genet. 2014;15:14.
Do DN, Strathe AB, Ostersen T, Pant SD, Kadarmideen HN. Genome-wide association and pathway analysis of feed efficiency in pigs reveal candidate genes and pathways for residual feed intake. Front Genet. 2014;5:307.
Jeong KI, Suzuki H, Nakayama H, Doi K. Ultrastructural study on the follicle-associated epithelium of nasal-associated lymphoid tissue in specific pathogen-free (SPF) and conventional environment-adapted (SPF-CV) rats. J Anat. 2000;196(3):443–51.
Yang D, Zhao C, Zhang M, Zhang S, Zhai J, Gao X, et al. Changes in oxidation-antioxidation function on the thymus of chickens infected with reticuloendotheliosis virus. BMC Vet Res. 2020;16(1):483.
Itoh K, Maejima K, Ueda K, Fujiwara K. Difference in susceptibility of mice raised under barrier-sustained (SPF) or conventional conditions to infectious megaenteron. Microbiol Immunol. 1979;23(9):909–13.
Wlasiuk G, Khan S, Switzer WM, Nachman MW. A history of recurrent positive selection at the toll-like receptor 5 in primates. Mol Biol Evol. 2009;26(4):937–49.
Chapman JR, Hellgren O, Helin AS, Kraus RH, Cromie RL, Waldenström J. The evolution of Innate Immune genes: purifying and balancing selection on β-Defensins in Waterfowl. Mol Biol Evol. 2016;33(12):3075–87.
Hu XJ, Yang J, **e XL, Lv FH, Cao YH, Li WR, et al. The genome landscape of tibetan sheep reveals adaptive introgression from Argali and the history of early human settlements on the Qinghai-Tibetan Plateau. Mol Biol Evol. 2019;36(2):283–303.
Zhong HA, Kong XY, Zhang YW, Su YK, Zhang B, Zhu L, et al. Microevolutionary mechanism of high-altitude adaptation in tibetan chicken populations from an elevation gradient. Evol Appl. 2022;15(12):2100–12.
Dodgson JB, Delany ME, Cheng HH. Poultry genome sequences: progress and outstanding challenges. Cytogenet Genome Res. 2011;134(1):19–26.
Zhang S, Jiang E, Wang K, Zhang Y, Yan H, Qu L, et al. Two insertion/deletion variants within SPAG17 gene are associated with goat body measurement traits. Animals. 2019;9(6):379.
van der Valk RJ, Kreiner-Møller E, Kooijman MN, Guxens M, Stergiakouli E, Sääf A, et al. A novel common variant in DCST2 is associated with length in early life and height in adulthood. Hum Mol Genet. 2015;24(4):1155–68.
Zhao J, Li M, Bradfield JP, Zhang H, Mentch FD, Wang K, et al. The role of height-associated loci identified in genome wide association studies in the determination of pediatric stature. BMC Med Genet. 2010;11:96.
Kim JJ, Lee HI, Park T, Kim K, Lee JE, Cho NH, et al. Identification of 15 loci influencing height in a Korean population. J Hum Genet. 2015;55(1):27–31.
Guo X, Pei J, Wu X, Bao P, Ding X, **ong L, et al. Detection of InDel and CNV of SPAG17 gene and their associations with bovine growth traits. Anim Biotechnol. 2002;33(3):440–7.
Jüppner H. Jansen’s metaphyseal chondrodysplasia: a disorder due to a PTH/PTHrP receptor gene mutation. Trends Endocrinol Metab. 1996;7(5):157–62.
Ren T, Zhang Z, Fu R, Yang Y, Li W, Liang J, et al. A 51 bp indel polymorphism within the PTH1R gene is significantly associated with chicken growth and carcass traits. Anim Genet. 2020;51(4):568–78.
Zheng L, Xu J, Liu X, Zhang Z, Zhong J, Wen Y, et al. The copy number variation of DMBT1 gene effects body traits in two Chinese cattle breeds. 3 Biotech. 2022;12(4):93.
Sun Y, Shi N, Lu H, Zhang J, Ma Y, Qiao Y, et al. ABCC4 copy number variation is associated with susceptibility to esophageal squamous cell carcinoma. Carcinogenesis. 2014;35(9):1941–50.
Braff MH, Bardan A, Nizet V, Gallo RL. Cutaneous defense mechanisms by antimicrobial peptides. J Invest Dermatol. 2005;125(1):9–13.
Pranjol MZI, Zinovkin DA, Maskell ART, Stephens LJ, Achinovich SL, Los’ DM, et al. Cathepsin L-induced galectin-1 may act as a proangiogenic factor in the Metastasis of high-grade serous carcinoma. J Transl Med. 2019;17(1):216.
Liao X, Peng F, Forni S, McLaren D, Plastow G, Stothard P. Whole genome sequencing of Gir cattle for identifying polymorphisms and loci under selection. Genome. 2013;56(10):592–8.
Haase B, Humphray SJ, Lyer S, Renner M, Poustka A, Mollenhauer J, et al. Molecular characterization of the porcine deleted in malignant brain tumors 1 gene (DMBT1). Gene. 2006;376(2):184–91.
Caterina MJ. Transient receptor potential ion channels as participants in thermosensation and thermoregulation. Am J Physiol Regul Integr Comp Physiol. 2007;292(1):R64–R76.
Zhou Q, Fu X, Xu J, Dong S, Liu C, Cheng D, et al. Hypothalamic warm-sensitive neurons require TRPC4 channel for detecting internal warmth and regulating body temperature in mice. Neuron. 2022;111(3):387–404e8.
Howard JT, Kachman SD, Snelling WM, Pollak EJ, Ciobanu DC, Kuehn LA, et al. Beef cattle body temperature during climatic stress: a genome-wide association study. Int J Biometeorol. 2014;58(7):1665–72.
Sinding MS, Gopalakrishnan S, Ramos-Madrigal J, de Manuel M, Pitulko VV, Kuderna L, et al. Arctic-adapted dogs emerged at the Pleistocene-Holocene transition. Science. 2020;368(6498):1495–9.
Wang YN, Tang Y, He Z, Ma H, Wang L, Liu Y, et al. Slit3 secreted from M2-like macrophages increases sympathetic activity and thermogenesis in adipose tissue. Nat Metab. 2021;3(11):1536–51.
Siegler RW. Fatal heatstroke in a young woman with previously undiagnosed Hashimoto’s thyroiditis. J Forensic Sci. 1998;43(6):1237–40.
Burger MF, Denver RJ. Plasma thyroid hormone concentrations in a wintering passerine bird: their relationship to geographic variation, environmental factors, metabolic rate, and body fat. Physiol Biochem Zool. 2002;75(2):187–99.
**e S, Yang X, Wang D, Zhu F, Yang N, Hou Z, et al. Thyroid transcriptome analysis reveals different adaptive responses to cold environmental conditions between two chicken breeds. PLoS ONE. 2018;13(1):e0191096.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Key R&D Program of China (2021YFF0703000), the National Natural Science Foundation of China (31872313), grants from the State Key Laboratory of Veterinary Biotechnology Program (SKLVBP202101, SKLVBP202120), and the Central Public- interest Scientific Institution Basal Research Fund (1610302022018).
Author information
Authors and Affiliations
Contributions
LL and JQ conceived and designed the experiments. LL and HL performed the experiments. HY and HC analyzed the data. CX and SZ contributed reagents, materials and tools and collected the samples. LL and CG wrote the manuscript and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The animals and experimental methods were performed according to the Chinese Legislation on the Use and Care of Laboratory Animals and were approved by the ethical review board of Harbin Veterinary Research Institute of the Chinese Academy of Agricultural Sciences (Approval No.: Heilongjiang SYXK-2006-032). The authors declare that all animal experiments and methods were performed in accordance with the relevant guidelines and followed the ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.


12864_2023_9928_MOESM10_ESM.xls
Additional file 10: Table S8. Kyoto Encyclopedia of Genes and Genomes (KEGG) functional enrichment of CNVR-harbored genes
12864_2023_9928_MOESM12_ESM.xls
Additional file 12: Table S10. KEGG analysis of CNVR-harbored that are genes differentially expressed in three duck populations
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Li, L., Quan, J., Liu, H. et al. Identification of the genetic characteristics of copy number variations in experimental specific pathogen-free ducks using whole-genome resequencing. BMC Genomics 25, 17 (2024). https://doi.org/10.1186/s12864-023-09928-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09928-8