
Abstract
Backgroud
The single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) are two major genomic variants, which play crucial roles in evolutionary and phenotypic diversity.
Results
In this study, we performed a comprehensive analysis to explore the genetic variations (SNPs and CNVs) of high sperm motility (HSM) and poor sperm motility (PSM) Simmental bulls using the high-coverage (25×) short-read next generation sequencing and single-molecule long reads sequencing data. A total of ~ 15 million SNPs and 2,944 CNV regions (CNVRs) were detected in Simmental bulls, and a set of positive selected genes (PSGs) and CNVRs were found to be overlapped with quantitative trait loci (QTLs) involving immunity, muscle development, reproduction, etc. In addition, we detected two new variants in LEPR, which may be related to the artificial breeding to improve important economic traits. Moreover, a set of genes and pathways functionally related to male fertility were identified. Remarkably, a CNV on SPAG16 (chr2:101,427,468 − 101,429,883) was completely deleted in all poor sperm motility (PSM) bulls and half of the bulls in high sperm motility (HSM), which may play a crucial role in the bull-fertility.
Conclusions
In conclusion, this study provides a valuable genetic variation resource for the cattle breeding and selection programs.
Similar content being viewed by others

Backgroud
Cattle were domesticated around 10,000 years before present (YBP), providing mankind with meat, milk, skin, and working power, etc. The natural and artificial selection has left phenomenal stress marks on the cattle genome determining its phenotype, adaptation and production performance. To date, a large number of studies have been reported in various cattle populations based on the genomic single nucleotide polymorphisms (SNPs), and a set of candidate genes were identified to be related to reproduction, meat, milk and environmental adaption. For instance, three genes (MATR3, MZB1 and STING1) are related to host immune, and SOD1, PRLH and DNAJC18 genes are associated with environmental thermal stress in the African cattle [1, 2]. Significantly, hundreds of candidate regions under positive selection among different cattle breeds were detected, which were responsible for production, growth, reproduction, immune response and milk production [3, 4].
Copy number variation (CNV) is another kind of genomic variant, ranging from 50 bp to 5 Mb [5]. Compared to the SNPs, CNV has a greater influence on the function, phenotype and evolution by the gene dosage, coding sequence, and regulation of long-range genes [6, 7]. The CNV in many species has been investigated based on comparative genomic hybridization arrays (CGH array), SNP arrays (Illumina BovineHD BeadChip, Illumina BovineSNP50 BeadChip), short-read next-generation sequencing (NGS) and single-molecule long-read sequencing (SMRT) methods. Compared to array technologies, NGS and SMRT exhibit higher precise breakpoints, sensitivity and resolution [8, 9]. As for the short-reads NGS data, many detection methods have been developed according to four strategies: read pair (RP), split read (SR), read depth (RD) and genome-based assembly (AS) [10]. Each strategy has its own strengths and weaknesses, and none of them can detect all types of CNVs. The SMRT can substantially improve the reliability and resolution of variant detection [11]. However, due to the high cost, limited studies were conducted to detect CNV in cattle genome using SMRT.
The Simmental cattle, a beef/milk dual-purpose breed, is one of the most widely distributed cattle breeds in the world. Previous studies have explored the selective signatures and copy number variation in Simmental cattle using different SNP arrays. The selection signature was firstly investigated in a large population of Simmental cattle using Illumina BovineSNP50, which identified 224 candidate regions containing genes associated with important economical traits [12]. Another study identified 263 CNV regions (CNVRs) in the genome of Simmental cattle using Illumina Bovine HD BeadChip, revealing that genes in CNVRs are related to transmembrane activity and olfactory transduction activity [13]. Afterward, various genome-wide association studies (GWAS) have been performed to identify the candidate genes/loci associated with economic traits of Simmental cattle, including carcass, meat, and growth [14,15,16]. Although it is feasible to detect the positive selective signatures and CNV using the SNP array, the limited resolution reduces the sensitivity and accuracy of detection. In addition, fertility plays an important role in the success of calf production. To date, most studies primarily focused on the female fertility, while male fertility has received much less attention. Though previous studies have identified a set of single-nucleotide polymorphisms (SNPs) associated with bull fertility based on the SNP array [16,17,18,19], the effective markers are still lacking for elite bull selection.
In the current study, combining the high-coverage short-read NGS data and SMRT data, a comprehensive analysis was conducted to identify the genetic variations (SNPs and CNVs) in the genome of Simmental bulls, which identified a set of positive selected genes (PSGs) and CNVRs overlapped with quantitative trait loci (QTLs) involving in milk, immunity, reproduction. Significantly, a CNV on SPAG16 was completely deleted in all poor sperm motility (PSM) bulls and half of the high sperm motility (HSM) bulls, indicating its important role in bull fertility.
Results
Genomic landscape of SNPs and CNVs in Simmental bulls
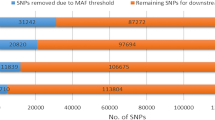
In the present study, 30 Simmental cattle were sequenced generating ~ 2.17 billion paired-end reads with the average coverage depth of ~ 25×. The reads were aligned to the high-quality taurine reference genome (ARS-UCD1.2) [20] with an average alignment rate of 99.83% (Table S1), generating 15,154,539 autosomal SNPs (121,568 with a minor allele frequency < 1%, 2,643,818 between 1% and 5%, and 11,490,159 > 5%) which were used for further downstream analysis.
In addition, we constructed a confidential CNV dataset using high-throughput Nanopore long-reads (PromethION) and the high coverage Illumina short read sequencing data. A total of 2,944 copy number variant regions (CNVRs) were detected (Fig. 1a &c, Table S2), including 1,651 deletions, 126 duplications, 1,167 both events, with a total length of 4,661,581 bp and an average length of 1,583 bp, covering 0.18% of the genome. The length of CNVRs was found to be mainly distributed within 100–500 bp, accounting for ~ 43.65% of the detected CNVR (Fig. 1b, Table S3). Moreover, with the increase in its length (> 500 bp), the number of CNVRs decreases. In addition, our results showed that CNVRs were not uniformly distributed across the genome, of which ~ 60.53% of CNVRs (1,782) were located in the intergenic region, only 0.71% in the exon region (Fig. 1d). Moreover, there were 11 CNVRs overlapped with the QTLs related to immunity, milk and production traits (Table S4).

(a) Genomic distribution of CNVRs. Green: both, red: deletion, blue: duplication. (b) Frequency of different types of CNVR (c) CNVR length distribution. (d) Functional classification of the detected CNVRs
Selection signature analysis for Simmental cattle based on autosomal SNPs
Three statistical methods (Pi, CLR, and iHS) (Table S5-S7) were applied to explore the positive selection signatures in Simmental cattle. For each method, regions showing outlier values (top 1%) were selected as the candidate genomic regions (Figure S2a). The positive selected genes (PSGs) were identified by at least two approaches, and a total of 235 PSGs were identified.
Totally, 53 PSGs were intersected with cattle QTLs (containing 476 QTLs) [21], which were associated with immunity, meat, milk, production, and reproduction (Table S8). For example, a region on chromosome 16 containing RERE and its neighboring genes (LOC112441839 and SLC45A1) was observed with markedly higher values, falling in 4 QTLs associated with reproduction (Figure S2 a&b, Table S8). In addition, another ~ 2.5 Mb region on chromosome 7 also showed markedly higher values, containing ten genes (ANKHD1, CDC23, CXXC5, CYSTM1, FAM13B, KDM3B, LOC101904825, NRG2, PSD2, PURA), overlapped with QTLs associated with immunity, milk and reproduction in cattle (Figure S2 a&c, Table S8).
Moreover, KOBAS [22] was used to perform GO and KEGG pathway analysis based on the 235 PSGs (Table S9-S10). The KEGG pathways resulted in two significantly over-represented pathways: cytokine-cytokine receptor interaction (corrected P-Value = 0.0159) and Oocyte meiosis (corrected P-Value = 0.0177). A total of 8 genes (TNFSF13, TNFSF12, LEPR, IFNAR2, EDAR, CX3CL1, GDF15, CCL22) were involved in the cytokine-cytokine receptor interaction pathway. Notably, two conserved nonsynonymous mutations (rs43347904, g.79,817,216: G > A, exon3, p.S6F; rs43347906, g.79,817,216: C > A, exon4, p.V35L) were detected within the LEPR gene (Fig. 2). LEPR was detected within a QTL related to reproduction in cattle [21]. We further investigated the frequency of these two mutations across the diverse cattle breeds around the world using the Bovine Genome Variation Database and Selective Signatures (BGVD, http://animal.nwsuaf.edu.cn/code/index.php/BosVar) (Fig. 2c and d). The allele G of rs43347904 showed a high frequency in European (0.724) and Eurasian (0.789) cattle populations. In Simmental cattle populations, the allele G showed a higher frequency of 0.957. It was also observed in some Chinese and African cattle breeds with a low frequency. The rs43347906 showed a similar pattern, and alters the protein structure (Fig. 2e). In addition, by investigating published literatures, we found a set of genes associated with immunity (EGR1, MUC6), and muscle development (MEIS1, GDF15) (Table 1).

Selection and conservative analysis of LEPR. (a) Selective sweep detected by Tajima’s D and Pi test. (b) conservative analysis of two loci. (c & d) Geographical distribution of the two variants (http://animal.nwsuaf.edu.cn/code/index.php/BosVar) (e) 3D protein structure prediction of LEPR
Identification of candidate genes/CNVRs associated with fertility
Sperm motility is one of the major determinants of male fertility. According to the sperm motility, the 30 Simmental bulls were divided into two groups: the HSM (n = 14) and the PSM group (n = 16). For HSM group, the sperm motility of fresh semen and frozen-thawed semen were 0.68 ± 0.04, and 0.36 ± 0.02, respectively, while were 0.32 ± 0.11 and 0.15 ± 0.05 for PSM group, respectively. To investigate the group-specific selection between HSM and PSM groups, we scan for genomic regions from genome-wide SNP and CNV datasets, respectively.
Firstly, we used the FST to scan genomic regions with extreme allele frequency differentiation between HSM and PSM groups using the SNP dataset. Using the top 1% of FST values, we identified 564 candidate selective regions covering 599 genes. To obtain a broad overview of the molecular functions of these candidate genes, we performed GO and KEGG enrichment analysis using KOBAS [22]. We detected three significantly over-represented (corrected P-value < 0.05) KEGG pathways related to the male fertility (insulin secretion (corrected P-value = 0.01537), oxytocin signaling pathway (corrected P-value = 0.016431), calcium signaling pathway (corrected P-value = 0.02558)) (Table S11). Besides, we identified 13 significantly over-represented GO terms (Table S12), such as potassium ion transmembrane transport, stabilization of membrane potential, potassium ion leak channel activity, calcium ion binding, etc.
In addition, a comparison of these detected candidate regions and known QTLs revealed that these detected candidate genes are overlapped with cattle QTLs associated with reproduction, production, and health (Table S13). Totally, our results showed that there were 290 candidate genes were observed to overlap with 2,611 cattle QTLs (Table S13). Among these 2,611 cattle QTLs, 284 QTLs covering 101 genes (such as SERPINE2, AGBL4, SORCS1, TMEM181, SPAG16) were associated with reproduction traits, such as sperm motility (AGBL4, SORCS1, SPAG16), sperm concentration (TMEM181), and fertilization rate (SERPINE2).
Moreover, we identified 58 highly differentiated CNVRs (top 2% of VST value) between the two groups (Fig. 3a) by calculating VST based on the confidential CNV dataset constructed in this study. Our results showed that 26 CNVRs were found to be located in the intergenic region, while 21 lay in the intronic region, overlap** with 31 genes. Some of these genes (such as ARID4A, ALDH8A1, and SPAG16) were involved in the male fertility. We detected a significantly differential deletion (chr2:101427468–101,429,883) covering the intronic region of SPAG16 gene (Fig. 3b). We used the PCR to check the existence of this CNV segment in 30 Simmental cattle (Figure S3). The results showed that this region was a complete deletion in all the PSM bulls and half of the HSM bulls based on the genoty** information, which confirmed our observation in the genome sequencing analysis. In addition, the SPAG16 was also identified as a candidate gene in the FST. Moreover, the expression level of SPAG16 in cattle was significantly higher in testis than in other tissues (http://animal.nwsuaf.edu.cn/code/index.php/RGD/loadByGet?address[]=RGD/Items/ExprCattle), indicating that SPAG16 may play an important role in the male fertility of Simmental cattle (Fig. 3c).

(a) Manhattan plot for VST (top 2%). (b) Boxplot of normalized copy number of SPAG16. (c) Gene expression of SPAG16 in different cattle tissues (http://animal.nwsuaf.edu.cn/code/index.php/RGD)
Discussion
As one of the most important and widely distributed cattle breeds, Simmental cattle are mainly used for milk and beef purposes. In the current study, we detected 15,154,539 autosomal SNPs in Simmental cattle. Besides, we firstly constructed a confidential CNV data set for Simmental cattle using different sequencing platforms (NGS and long-reads), multi-strategies, and the newly reported high-quality genome (ARS-UCD1.2) [20], which ensure us to obtain a highly confidential CNV dataset. Totally, we detected a total of 2,944 CNVRs for 30 Simmental cattle, which showed a similar level to other the cattle breeds [23]. Compared with cattle QTL database, there were 11 CNVRs overlapped with QTLs related to immunity, milk and production traits [21], indicating that CNV may be a critical type of genetic variation, may have an important effect on cattle fertility, health, and economic traits.
Over the last few decades, strong human driven selection contributed immensely to productive traits enhancement within the Simmental cattle genome. It is worthwhile to identify the candidate gene during the domestication, which will accelerate the improvement of important traits of cattle in the future. In this study, we used three methods (Pi, CLR, and iHS) to improve the power of detecting selection signatures [24], and a total of 235 PSGs were identified for Simmental cattle. To further explore the hereditary effects, the detected PSGs were compared with cattle QTLs [21]. A total of 53 PSGs overlapped with 469 QTLs related to immunity, meat, milk, production, and reproduction [21] (Table S1, Table S2). Notably, a ~ 2.5 Mb region on chromosome 7 containing several genes (ANKHD1, CDC23, CXXC5, CYSTM1, FAM13B, KDM3B, LOC101904825, NRG2, PSD2, PURA) and another region on chromosome 16 containing RERE gene and its neighboring genes (LOC112441839 and SLC45A1) showing high values, was overlapped with QTLs associated with milk and reproduction traits in cattle [21]. Studies showed that CYSTM1 and NRG2 are significantly related to the gestation length [25, 26]. KDM3B, a histone H3 demethylase, plays a crucial role in spermatogenesis and normal male sexual behavior, which is also identified as a fertility-related candidate gene for sheep [27]. CXXC5 and PSD2 were involved in the function of fat deposition [28, 29]. The RERE has been identified as a candidate gene associated with reproductive development [30,31,32]. The functional analysis (KEGG pathway and GO) performed based on the 235 PSGs showed that cytokine-cytokine receptor interaction (corrected P-Value = 0.0159) and oocyte meiosis (corrected P-Value = 0.0177) were significantly over-represented. The oocyte meiosis pathway has been reported to be important in reproduction [33, 34]. The cytokine-cytokine receptor interaction pathway plays a central role in immunity, which is also related to backfat thickness [35], reproduction [34], growth of the animal [36], feed conversion ratio in beef cattle [37], beef quality [62]. A study reported that ARID4A was associated with the semen quality of bulls [63]. ALDH8A1 can synthesize retinoic acid which plays an important role during spermatogenesis [64, 65]. SPAG16, plays an important role in spermatogenesis [60, 61]. In addition, the expression level of SPAG16 was significantly higher in the cattle testis, compared to other organs (http://animal.nwsuaf.edu.cn/code/index.php/RGD). Notably, a differential deletion (chr2:101427468–101,429,883) was detected following the intronic region of SPAG16 gene, which was mainly observed in bulls of the PSM group. Meantime, half of the HSM bulls showed a loss of heterozygosity. Moreover, the SPAG16 was also identified as a candidate gene associated with reproduction by FST. Therefore, we speculated that SPAG16 plays a crucial role in the male fertility.
Conclusions
In the current study, we performed a comprehensive analysis to explore the genetic variations (SNPs and CNVs) in Simmental cattle. We identified a set of candidate genes associated with reproduction, immunity, milk, and muscle development. In addition, we obtained a confidential CNV dataset and sperm-motility-related CNVRs genes for Simmental cattle using the high-coverage next-generation re-sequencing and long read sequencing. We admitted that this CNV dataset we constructed is not fully complete due to the strict filtering standards, and limited sample. In future research, combining various sequencing platforms and improved detection methods, we may obtain an infinitely close to complete dataset.
Methods
Sample collection and genomic sequencing
The frozen semen of 30 Simmental cattle were obtained from Gansu Livestock Breeding Center (Gansu Province, China), which can be divided into high sperm motility (HSM) (n = 14) and poor sperm motility (PSM) (n = 16) based on sperm motility. The sperm motility of fresh semen and frozen-thawed semen were calculated using Minitube Sperm Vision for each individual with at least five ejaculations (CASA SpermVision®, Minitube, Germany). The genomic DNA was extracted using a standard phenol-chloroform protocol [66]. High-quality DNA was processed to construct the short-insert (500 bp) genomic libraries on BGISEQ-500 for genome sequencing (BGI Biotech Co. Ltd, Bei**g, China). In addition, one out of 30 bulls was randomly selected for single-molecule long-read sequencing by Nanopore PromethION Platform (Nextomics Biosciences Co., Ltd, Wuhan, China).
Alignments and variant identification
The cleaned reads of 30 bulls were aligned to the latest high quality reference genome (ARS-UCD1.2) [20] using BWA-MEM with default settings [67]. Duplicate reads were filtered using Picard (v2.5.0). The single nucleotide polymorphisms (SNPs) were detected with the Genome Analysis Toolkit (GATK, version 3.8) [68]. All SNPs were filtered using the “VariantFiltration” implemented in GATK with the standards used in the previous studies [69]. In addition, the Nanopore long reads were mapped to the cattle genome (ARS-UCD1.2) using minimap2 with default settings [70].
Genome-wide selective sweep tests
The positive genomic regions in Simmental bulls were estimated using three statistical methods, including nucleotide diversity (Pi), integrated Haplotype Score (iHS) and composite likelihood ratio (CLR). The Pi was calculated with 50 kb sliding windows and 20 kb steps along the autosomes using the vcftools. The iHS based on the phased genotype data was performed using selscan v1.1, and score was normalized with the norm module with 50 kb windows and 20 kb increments [71]. The CLR was calculated by SweepFinder2 with each 50-kb window across each chromosome [72]. The KOBAS (http://kobas.cbi.pku.edu.cn/) [22] was used to gain a better understanding of the biological functions and involved pathways. The 3D structures of LEPR was predicted using I-TASSER (https://zhanglab.ccmb.med.umich.edu/I-TASSER/)[73], which were visualized using UCSF Chimera [74].
CNV identification
Sniffles (Version: 1.0.10) was used to detect structure variation (SV) based on the Nanopore long reads with default parameter [75]. SV analysis outputs were filtered with the following three steps: (1) ambiguous breakpoints (flag: IMPRECISE) and low-quality SV were removed; (2) SVs shorter than 50 bp were removed; (3) SVs with less than four supporting reads were removed. Lumpy (v 0.2.13) was performed for each sample to detect the read-pair and split-read profile CNV call set using the lumpyexpress module with default parameters [76]. CNVnator was used to annotate the copy number [77]. The CNVs were identified as the same type by the three methods to ensure confidence. After considering the intersections between the results of Sniffles, LUMPY and CNVnator, only CNVRs supported by at least two animals were kept (Table S1).
Detection of candidate genes/CNVRs associated with fertility
The FST was used to scan genomic regions with extreme allele frequency differentiation between HSM and PSM groups using the SNP dataset with 50 kb sliding windows and 20 kb steps along the autosomes by vcftools (0.1.16) [78]. A custom script was used to calculate the VST using the identified CNVR data set [79]. The formula is VST = (VT - VS)/VT, where VT represents the variance apparent among all unrelated individuals, and VS represents the average variance within each population, weighted for population size. The top 2% VST were considered to have a significant difference in copy number between the fertile group and sub-fertile group.
Validation of a deletion within SPAG16 by PCR
The deletion at SPAG16 (chr2:101,427,468 − 101,429,883) was verified with primers F: 5’-CATGAGGATCAGTGCTGCTG-3’ and R: 5’-GGCACTTCCTTGATCCACACA − 3’. The polymerase chain reaction (PCR) system contained 12.5 µL of 2× EasyTaq PCR SuperMix Polymerase (TransGen Biotech, Bei**g, China), 50 ng of genomic DNA, 1 µL of each primer (0.1 µmol/µL), and then adding distilled water to a total volume of 25 µL. The amplification conditions were pre-denaturation at 94 °C for 5 min, followed by 34 cycles of denaturation at 94 °C for 30 s, annealing for 30 s, 72 °C extension for 30 s, and a final extension at 72 °C for 5 min. The PCR products were detected by gel electrophoresis and visualized under UV illumination (Ge1Doc-It TS Imaging System, Upland, CA, USA).
Functional annotation
In order to explore the biological function and pathway of the identified positive selected genes and CNVR genes, the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway and Gene Ontology (GO) were performed using KOBAS with a significant threshold for corrected P-value < 0.05 [22]. The cattle quantitative trait loci (QTLs) data were obtained from Animal QTLdb [21]. All QTL data were filtered with P ≤ 0.05. To further explore the hereditary effects, the positive selected genes and CNVRs were compared with cattle QTLs using Bedtools with the parameters: -a -b -r -wa -wb [80].
Data availability
The raw data have been deposited to NCBI with the BioProject accession number PRJNA701080 and PRJNA950231.
Abbreviations
- BGV:
-
Bovine genome variation database and selective signatures
- CGH:
-
Comparative genomic hybridization
- CLR:
-
Composite likelihood ratio
- CNV:
-
Copy number variation
- CNVR:
-
CNV region, YBP:Years before present
- GATK:
-
Genome Analysis Toolkit
- GWAS:
-
Genome-wide association study
- HSM:
-
High sperm motility
- iHS:
-
Integrated haplotype Score
- Pi:
-
Nucleotide diversity
- PSG:
-
Positive selected genes
- PSM:
-
Poor sperm motility
- QTLs:
-
Quantitative trait loci
- RP:
-
Read pair, SR:Split read
- RD:
-
Read depth
- SNP:
-
Single nucleotide polymorphisms
- SV:
-
Structure variation
References
Kim J, Hanotte O, Mwai OA, Dessie T, Bashir S, Diallo B, Agaba M, Kim K, Kwak W, Sung S, et al. The genome landscape of indigenous african cattle. Genome Biol. 2017;18(1):34.
Kim K, Kwon T, Dessie T, Yoo D, Mwai OA, Jang J, Sung S, Lee S, Salim B, Jung J, et al. The mosaic genome of indigenous african cattle as a unique genetic resource for african pastoralism. Nat Genet. 2020;52(10):1099–110.
Chen M, Pan D, Ren H, Fu J, Li J, Su G, Wang A, Jiang L, Zhang Q, Liu J-F. Identification of selective sweeps reveals divergent selection between chinese holstein and simmental cattle populations. Genet Selection Evol. 2016;48(1):76.
Xu L, Bickhart DM, Cole JB, Schroeder SG, Song J, Tassell CPV, Sonstegard TS, Liu GE. Genomic signatures reveal new evidences for selection of important traits in domestic cattle. Mol Biol Evol. 2015;32(3):711–25.
Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK. Map** copy number variation by population-scale genome sequencing. Nature. 2011;470(7332):59–65.
Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8(8):639–46.
Henrichsen CN, Chaignat E, Reymond A. Copy number variants, diseases and gene expression. Hum Mol Genet. 2009;18(R1):R1–R8.
Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20(1):117.
Ho SS, Urban AE, Mills RE. Structural variation in the sequencing era. Nat Rev Genet. 2020;21(3):171–89.
Zhao M, Wang Q, Wang Q, Jia P, Zhao Z. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives. BMC Bioinformatics. 2013;14(11):1.
Jenko Bizjan B, Katsila T, Tesovnik T, Šket R, Debeljak M, Matsoukas MT, Kovač J. Challenges in identifying large germline structural variants for clinical use by long read sequencing. Comput Struct Biotechnol J. 2020;18:83–92.
Fan H, Wu Y, Qi X, Zhang J, Li J, Gao X, Zhang L, Li J, Gao H. Genome-wide detection of selective signatures in Simmental cattle. J Appl Genet. 2014;55(3):343–51.
Wu Y, Fan H, **g S, **a J, Chen Y, Zhang L, Gao X, Li J, Gao H, Ren H. A genome-wide scan for copy number variations using high-density single nucleotide polymorphism array in Simmental cattle. Anim Genet. 2015;46(3):289–98.
**a J, Qi X, Wu Y, Zhu B, Xu L, Zhang L, Gao X, Chen Y, Li J, Gao H. Genome-wide association study identifies loci and candidate genes for meat quality traits in simmental beef cattle. Mamm Genome. 2016;27(5):246–55.
Zhu B, Niu H, Zhang W, Wang Z, Liang Y, Guan L, Guo P, Chen Y, Zhang L, Guo Y, et al. Genome wide association study and genomic prediction for fatty acid composition in chinese simmental beef cattle using high density SNP array. BMC Genomics. 2017;18(1):464.
**a J, Fan H, Chang T, Xu L, Zhang W, Song Y, Zhu B, Zhang L, Gao X, Chen Y, et al. Searching for new loci and candidate genes for economically important traits through gene-based association analysis of Simmental cattle. Sci Rep. 2017;7:42048.
Sweett H, Fonseca PAS, Suárez-Vega A, Livernois A, Miglior F, Cánovas A. Genome-wide association study to identify genomic regions and positional candidate genes associated with male fertility in beef cattle. Sci Rep. 2020;10(1):20102.
Han Y, Peñagaricano F. Unravelling the genomic architecture of bull fertility in Holstein cattle. BMC Genet. 2016;17(1):143.
Peñagaricano F, Weigel KA, Khatib H. Genome-wide association study identifies candidate markers for bull fertility in Holstein dairy cattle. Anim Genet. 2012;43(s1):65–71.
Rosen BD, Bickhart DM, Schnabel RD, Koren S, Elsik CG, Tseng E, Rowan TN, Low WY, Zimin A, Couldrey C, et al. De novo assembly of the cattle reference genome with single-molecule sequencing. GigaScience. 2020;9(3):giaa021.
Hu Z-L, Park CA, Reecy JM. Building a livestock genetic and genomic information knowledgebase through integrative developments of animal QTLdb and CorrDB. Nucleic Acids Res. 2019;47(D1):D701–10.
Bu D, Luo H, Huo P, Wang Z, Zhang S, He Z, Wu Y, Zhao L, Liu J, Guo J, et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021;49(W1):W317–25.
Mei C, Junjvlieke Z, Raza SHA, Wang H, Cheng G, Zhao C, Zhu W, Zan L. Copy number variation detection in chinese indigenous cattle by whole genome sequencing. Genomics. 2020;112(1):831–6.
Zeng K, Shi S, Wu C-I. Compound tests for the detection of hitchhiking under positive selection. Mol Biol Evol. 2007;24(8):1898–908.
Fang L, Jiang J, Li B, Zhou Y, Freebern E, Vanraden PM, Cole JB, Liu GE, Ma L. Genetic and epigenetic architecture of paternal origin contribute to gestation length in cattle. Commun Biology. 2019;2(1):100.
Purfield DC, Evans RD, Carthy TR, Berry DP. Genomic Regions Associated With Gestation Length Detected Using Whole-Genome Sequence Data Differ Between Dairy and Beef Cattle. Frontiers in Genetics 2019, 10(1068).
Dolebo AT, Khayatzadeh N, Melesse A, Wragg D, Rekik M, Haile A, Rischkowsky B, Rothschild MF, Mwacharo JM. Genome-wide scans identify known and novel regions associated with prolificacy and reproduction traits in a sub-saharan african indigenous sheep (Ovis aries). Mamm Genome. 2019;30(11):339–52.
Mastrangelo S, Bahbahani H, Moioli B, Ahbara A, Al Abri M, Almathen F, da Silva A, Belabdi I, Portolano B, Mwacharo JM, et al. Novel and known signals of selection for fat deposition in domestic sheep breeds from Africa and Eurasia. PLoS ONE. 2019;14(6):e0209632.
Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, Powell C, Vedantam S, Buchkovich ML, Yang J, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518(7538):197–206.
Ramey HR, Decker JE, McKay SD, Rolf MM, Schnabel RD, Taylor JF. Detection of selective sweeps in cattle using genome-wide SNP data. BMC Genomics. 2013;14(1):382.
Guarini AR, Lourenco DAL, Brito LF, Sargolzaei M, Baes CF, Miglior F, Misztal I, Schenkel FS. Genetics and genomics of reproductive disorders in canadian holstein cattle. J Dairy Sci. 2019;102(2):1341–53.
Liu Z, Oyola MG, Zhou S, Chen X, Liao L, Tien JC-Y, Mani SK, Xu J. Knockout of the histone demethylase Kdm3b decreases spermatogenesis and impairs male sexual behaviors. Int J Biol Sci. 2015;11(12):1447–57.
Celik O, Celik N, Gungor S, Haberal ET, Aydin S. Selective regulation of oocyte meiotic events enhances Progress in Fertility Preservation Methods. Biochem insights. 2015;8:11–21.
E G-X, Zhao Y-J, Huang Y-F. Selection signatures of litter size in Dazu black goats based on a whole genome sequencing mixed pools strategy. Mol Biol Rep. 2019;46(5):5517–23.
Mokry FB, Higa RH, de Alvarenga Mudadu M, Oliveira de Lima A, Meirelles SLC, Barbosa da Silva MVG, Cardoso FF, Morgado de Oliveira M, Urbinati I, Méo Niciura SC, et al. Genome-wide association study for backfat thickness in Canchim beef cattle using Random Forest approach. BMC Genet. 2013;14(1):47.
Chen B, Xu J, He X, Xu H, Li G, Du H, Nie Q, Zhang X. A genome-wide mRNA screen and functional analysis Reveal FOXO3 as a candidate gene for Chicken growth. PLoS ONE. 2015;10(9):e0137087.
de Almeida Santana MH, Junior GAO, Cesar ASM, Freua MC, da Costa Gomes R, da, Luz e Silva S, Leme PR, Fukumasu H, Carvalho ME, Ventura RV et al. Copy number variations and genome-wide associations reveal putative genes and metabolic pathways involved with the feed conversion ratio in beef cattle. Journal of Applied Genetics 2016, 57(4):495–504.
Guifen L, **aomu L, Fachun W, **uwen T, Haijian C, Enliang S. Use of a bovine genome chip to identify new biological pathways for beef quality in cattle. Mol Biol Rep. 2012;39(12):10979–86.
Fan H, Wu Y, Zhou X, **a J, Zhang W, Song Y, Liu F, Chen Y, Zhang L, Gao X, et al. Pathway-based genome-wide Association Studies for two meat production traits in Simmental cattle. Sci Rep. 2015;5(1):18389.
Li X, Yang J, Shen M, **e X-L, Liu G-J, Xu Y-X, Lv F-H, Yang H, Yang Y-L, Liu C-B, et al. Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nat Commun. 2020;11(1):2815.
Yang J, Li W-R, Lv F-H, He S-G, Tian S-L, Peng W-F, Sun Y-W, Zhao Y-X, Tu X-L, Zhang M, et al. Whole-genome sequencing of native Sheep provides insights into Rapid Adaptations to Extreme environments. Mol Biol Evol. 2016;33(10):2576–92.
Zhang J-X, Ma H-W, Sang M, Hu Y-S, Liang Z-N, Ai H-X, Zhang J, Cui X-W, Zhang S-Q. Molecular structure, expression, cell and tissue distribution, immune evolution and cell proliferation of the gene encoding bovine (Bos taurus) TNFSF13 (APRIL). Dev Comp Immunol. 2010;34(11):1199–208.
Zhang J-X, Sang M, Li J-F, Zhao W, Ma H-W, Min C, Hu Y-L, Du M-X, Zhang S-Q. Molecular structure and characterization of the cytokine TWEAK and its receptor Fn14 in bovine. Vet Immunol Immunopathol. 2011;144(3):238–46.
Jones BA, Beamer M, Ahmed S. Fractalkine/CX3CL1: a potential new target for inflammatory diseases. Mol Interv. 2010;10(5):263–70.
Widdison S, Coffey TJ. Cattle and chemokines: evidence for species-specific evolution of the bovine chemokine system. Anim Genet. 2011;42(4):341–53.
Rapp M, Wintergerst MWM, Kunz WG, Vetter VK, Knott MML, Lisowski D, Haubner S, Moder S, Thaler R, Eiber S, et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J Exp Med. 2019;216(5):1170–81.
Tsai VW, Macia L, Johnen H, Kuffner T, Manadhar R, Jorgensen SB, Lee-Ng KK, Zhang HP, Wu L, Marquis CP, et al. TGF-b superfamily cytokine MIC-1/GDF15 is a physiological appetite and body weight regulator. PLoS ONE. 2013;8(2):e55174.
Gurgul A, Szmatoła T, Ropka-Molik K, Jasielczuk I, Pawlina K, Semik E, Bugno-Poniewierska M. Identification of genome-wide selection signatures in the Limousin beef cattle breed. J Anim Breed Genet. 2016;133(4):264–76.
Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, Friedman JM. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269(5223):543–6.
Houseknecht KL, Portocarrero CP. Leptin and its receptors: regulators of whole-body energy homeostasis. Domest Anim Endocrinol. 1998;15(6):457–75.
Gorlov I, Sulimova G, Perchun A, Slozhenkina M. Genetic polymorphism of the RORC, BGH, BGHR, LEP, LEPR genes in Russian hornless cattle breed. In.; 2017.
Shi T, Xu Y, Yang M, Huang Y, Lan X, Lei C, Qi X, Yang X, Chen H. Copy number variations at LEPR gene locus associated with gene expression and phenotypic traits in chinese cattle. Anim Sci J. 2016;87(3):336–43.
Guo Y, Chen H, Lan X, Zhang B, Pan C, Zhang L, Zhang C, Zhao M. Novel SNPs of the bovine LEPR gene and their association with growth traits. Biochem Genet. 2008;46(11):828–34.
Szyndler-Nędza M, Tyra M, Ropka-Molik K, Piórkowska K, Mucha A, Różycki M, Koska M, Szulc K. Association between LEPR and MC4R genes polymorphisms and composition of milk from sows of dam line. Mol Biol Rep. 2013;40(7):4339–47.
Asadollahpour Nanaei H, Ansari Mahyari S, Edriss MA. Effect of LEPR, ABCG2 and SCD1 gene polymorphisms on Reproductive Traits in the iranian holstein cattle. Reprod Domest Anim. 2014;49(5):769–74.
Thackare H, Nicholson HD, Whittington K. Oxytocin—its role in male reproduction and new potential therapeutic uses. Hum Reprod Update. 2006;12(4):437–48.
Lu C-H, Lee RK-K, Hwu Y-M, Chu S-L, Chen Y-J, Chang W-C, Lin S-P, Li S-H. SERPINE2, a serine protease inhibitor extensively expressed in Adult Male Mouse Reproductive Tissues, May serve as a murine sperm decapacitation Factor1. Biol Reprod. 2011;84(3):514–25.
Li S-H, Hwu Y-M, Lu C-H, Lin M-H, Yeh L-Y, Lee RK-K. Serine protease inhibitor SERPINE2 Reversibly modulates murine sperm capacitation. Int J Mol Sci. 2018;19(5):1520.
Hering DM, Olenski K, Kaminski S. Genome-wide association study for poor sperm motility in Holstein-Friesian bulls. Anim Reprod Sci. 2014;146(3–4):89–97.
Zhang Z, Kostetskii I, Moss SB, Jones BH, Ho C, Wang H, Kishida T, Gerton GL, Radice GL, Strauss JF. Haploinsufficiency for the murine orthologue of Chlamydomonas PF20 disrupts spermatogenesis. Proc Natl Acad Sci USA. 2004;101(35):12946.
Nagarkatti-Gude DR, Jaimez R, Henderson SC, Teves ME, Zhang Z, Strauss JF. Spag16, an Axonemal Central Apparatus Gene, encodes a male germ Cell Nuclear speckle protein that regulates SPAG16 mRNA expression. PLoS ONE. 2011;6(5):e20625.
Wu R-C, Jiang M, Beaudet AL, Wu M-Y. ARID4A and ARID4B regulate male fertility, a functional link to the AR and RB pathways. Proceedings of the National Academy of Sciences 2013, 110(12):4616–4621.
Yang C, Wang J, Liu J, Sun Y, Guo Y, Jiang Q, Ju Z, Gao Q, Wang X, Huang J, et al. Functional haplotypes of ARID4A affect promoter activity and semen quality of bulls. Anim Reprod Sci. 2018;197:257–67.
Lin M, Napoli JL. cDNA cloning and expression of a human aldehyde dehydrogenase (ALDH) active with 9-cis-retinal and identification of a rat ortholog, ALDH12. J Biol Chem. 2000;275(51):40106–12.
Kent T, Griswold MD. Checking the Pulse of Vitamin A Metabolism and Signaling during Mammalian Spermatogenesis. Journal of Developmental Biology 2014, 2(1).
Green MR, Sambrook J. Molecular cloning: a Laboratory Manual (Fourth Edition): three-volume set. Cold Spring Harbor Laboratory Pr; 2012.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Nekrutenko A, Taylor J. Next-generation sequencing data interpretation: enhancing reproducibility and accessibility. Nat Rev Genet. 2012;13(9):667–72.
Chen N, Cai Y, Chen Q, Li R, Wang K, Huang Y, Hu S, Huang S, Zhang H, Zheng Z, et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat Commun. 2018;9(1):2337.
Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34(18):3094–100.
Szpiech ZA, Hernandez RD. selscan: an efficient Multithreaded Program to perform EHH-Based scans for positive selection. Mol Biol Evol. 2014;31(10):2824–7.
DeGiorgio M, Huber CD, Hubisz MJ, Hellmann I, Nielsen R. Sweep finder 2: increased sensitivity, robustness and flexibility. Bioinformatics. 2016;32(12):1895–7.
Yang J, Zhang Y. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res. 2015;43(W1):W174–181.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, Schatz MC. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15(6):461–8.
Layer RM, Chiang C, Quinlan AR, Hall IM. LUMPY: a probabilistic framework for structural variant discovery. Genome biology 2014, 15(6).
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21(6):974–84.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–8.
Huang Y, Li Y, Wang X, Yu J, Cai Y, Zheng Z, Li R, Zhang S, Chen N, Asadollahpour Nanaei H et al. An atlas of CNV maps in cattle, goat and sheep. Science China Life Sciences 2021:1–18.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–2.
Guo Y, Chen H, Lan X, Zhang B, Pan C, Zhang L, Zhang C, Zhao M. Novel SNPs of the bovine LEPR gene and their association with growth traits. Biochem Genet. 2008;46(11–12):828–34.
Szyndler-Nedza M, Tyra M, Ropka-Molik K, Piorkowska K, Mucha A, Rozycki M, Koska M, Szulc K. Association between LEPR and MC4R genes polymorphisms and composition of milk from sows of dam line. Mol Biol Rep. 2013;40(7):4339–47.
Rinaldi M, Dreesen L, Hoorens PR, Li RW, Claerebout E, Goddeeris B, Vercruysse J, Van Den Broek W, Geldhof P. Infection with the gastrointestinal nematode Ostertagia ostertagi in cattle affects mucus biosynthesis in the abomasum. Vet Res. 2011;42(1):61.
Zheng Z, Wang X, Li M, Li Y, Yang Z, Wang X, Pan X, Gong M, Zhang Y, Guo Y, et al. The origin of domestication genes in goats. Sci Adv. 2020;6(21):eaaz5216.
Grade CVC, Mantovani CS, Fontoura MA, Yusuf F, Brand-Saberi B, Alvares LE. CREB, NF-Y and MEIS1 conserved binding sites are essential to balance myostatin promoter/enhancer activity during early myogenesis. Mol Biol Rep. 2017;44(5):419–27.
Hou P, Zhao M, He W, He H, Wang H. Cellular microRNA bta-miR-2361 inhibits bovine herpesvirus 1 replication by directly targeting EGR1 gene. Vet Microbiol. 2019;233:174–83.
Acknowledgements
We would like to thank Dr. Zhuqing Zheng from Huazhong Agricultural University for his good suggestions.
Funding
The work was supported by National natural science foundation of China (31501918) to **angpeng Yue, the National Beef Cattle and Yak Industrial Technology System (CARS-37) to Chuzhao Lei, National natural science foundation of China (32200337) and China Postdoctoral Science Foundation (2022M712003) to Ting Sun.
Author information
Authors and Affiliations
Contributions
X.P.Y conceived and designed the study. T.S., S.W.P., Y.K.L performed statistical analysis. T.S. wrote the manuscript. Q.H., N.B.C., C.Z.L. revised the manuscript. H.Y.X. collected the samples. All authors reviewed the manuscript and gave final approval for publication.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All cattle and frozen sperm were handled following the guidelines in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of the People’s Republic of China. This study was approved by the Ethics Committee of the College of Pastoral Agriculture Science and Technology, Lanzhou University (Ethic approval No: 2010-1 and 2010-2).
This study was carried out in compliance with the ARRIVE guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sun, T., Pei, S., Liu, Y. et al. Whole genome sequencing of simmental cattle for SNP and CNV discovery. BMC Genomics 24, 179 (2023). https://doi.org/10.1186/s12864-023-09248-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-023-09248-x