
Abstract
Background
Merino sheep are the most famous fine wool sheep in the world. They have high wool production and excellent wool quality and have attracted worldwide attention. The fleece of the Merino sheep is composed predominantly of wool fibers grown from secondary wool follicles. Therefore, it is necessary to study the development of hair follicles to understand the mechanism of wool production. The hair follicle is a complex biological system involved in a dynamic process governed by gene regulation. The hair follicle development process is very complex and poorly understood. The purpose of our research is to identify candidate genes related to hair follicle development, provide a theoretical molecular breeding basis for the cultivation of fine wool sheep, and provide a reference for the problems of hair loss and alopecia areata that affect human beings.
Results
We analyzed mRNAs data in skin tissues of 18 Merino sheep at four embryonic days (E65, E85, E105 and E135) and two postnatal days (P7 and P30). G1 to G6 represent hair follicles developmental at six stages (i.e. E65 to P30). We identified 7879 differentially expressed genes (DEGs) and 12623 novel DEGs, revealed different expression patterns of these DEGs at six stages of hair follicle development, and demonstrated their complex interactions. DEGs with stage-specific expression were significantly enriched in epidermal differentiation and development, hair follicle development and hair follicle morphogenesis and were enriched in many pathways related to hair follicle development. The key genes (LAMA5, WNT10A, KRT25, SOSTDC1, ZDHHC21, FZD1, BMP7, LRP4, TGFβ2, TMEM79, SOX10, ITGB4, KRT14, ITGA6, and GLI2) affecting hair follicle morphogenesis were identified by network analysis.
Conclusion
This study provides a new reference for the molecular basis of hair follicle development and lays a foundation for further improving sheep hair follicle breeding. Candidate genes related to hair follicular development were found, which provided a theoretical basis for molecular breeding for the culture of fine wool sheep. These results are a valuable resource for biological investigations of fleece evolution in animals.
Similar content being viewed by others

Background
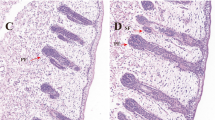
Subo Merino (SBM) is a superfine wool-producing sheep breed in China. The average wool fiber diameter is 17–19 µm, which surpasses the standard textile count of 80 Nm [1] and has a far-reaching impact on the fine wool sheep industry. The growth and development of sheep wool is controlled by hair follicles (HFs), which are tiny organs attached to the skin that have a complex morphology, complex structure and periodic growth [2]. HFs are composed of multiple cells with very intricate interactions and are involved in the regulation of HF development, growth, regeneration and differentiation. The development of wool follicles has been described in detail for the Merino and it is well established that no new follicles are initiated after birth [3,4,5,6,7]. The first stage in follicle development is the proliferation of epidermal cells to form a placode beneath which an aggregation of dermal cells occurs and the two cell formations grow down together into the dermis. Progressively, the dermal cells move into the epithelial bud to form the pre-papilla and finally the epithelial bulb cells envelop the pre-papilla as the follicle lengthens and descends into the dermis. The stages as they occur in Merino sheep is first follicles formed are the primary follicles (PFs) followed by secondary follicles (SFs) and then secondary-derived follicles (SD) that branch from the SFs [4]. In Merino sheep, fibres from the SD constitute the bulk of the fleece. The first follicles to be initiated in the sheep fetus PFs are visible from 75 days of gestation and are producing a fibre by 90 days of gestation [3]. SFs do not appear until approximately 85 days of gestation. Some of these follicles will begin to branch (SD) at around 105 days [8]. HFs fully mature after birth; therefore, the number of HFs does not increase after birth. In du Cros et al. [9] description of the localization of epidermal growth factor immunoreactivity in sheep skin during wool follicle development, it was found that immunoreactivity was restricted to the periderm and intermediate layers of fetal epidermis at 55 d of gestation, when the first wave of wool follicles are initiated. This particular distribution persisted during subsequent development but never became associated with the basal cells of the epidermis. At approximately 105 d of gestation, however, reactions were detected in the outer root sheath as the follicles matured and in the differentiating cells of the sebaceous glands. Hutchison and Mellor [6] study the effects of maternal nutrition on the initiation of secondary wool follicles in foetal Scottish Blackface sheep. They found initiation of SFs usually takes place between about 95 and 135 days of gestation. Severe underfeeding during the first half of this period did not significantly inhibit the initiation of SFs, but severe underfeeding during the latter half of this period resulted in a significantly lower number of SFs and this number was not increased by refeeding ewes to a high level between 132 days and term. They concluded that SFs initiation is most affected by maternal undernutrition between about 115 and 135 days.
The differentiation of HFs is regulated by a variety of signaling pathways, including the bone morphogenetic protein (BMP), transforming growth factor beta (TGF-β) and Wnt signaling pathways [10, 11]. The specific expression patterns of these molecules in dermal papillae or stromal cells determine their functions during differentiation. However, knowledge regarding the corresponding cellular and molecular mechanisms is limited. After the primary HFs form in the sheep fetus, branches of the primary HFs form secondary HFs [12, 13]. Therefore, it is important to understand the associated molecular gene regulation mechanisms. The wool quality and commercial value of fine wool sheep are determined by the structure and characteristics of their HFs. This branching can be extensive and determines the final follicle population density in which about 80% of the follicles are SD follicles and several wool fibres emerge at the skin surface from the same orifice [14]. To improve the wool yield and wool quality of these sheep, it is necessary to study the factors affecting the formation of HFs and to deeply understand the molecular regulatory mechanisms of HF development. The processes of HF cell development and differentiation are regulated by a variety of genes and multiple signaling pathways; thus, identifying the major genes regulating the development and differentiation of HF cells has become the focus of research.
Wool fiber fineness, fiber length, wool bending, wool strength and hair flexibility determine not only the differences between wool products and other textile fibers but also the craft value of wool textile products [24], GLI2, KRT25 [25, 26], LAMA5 [27, 28], LRP4 [29, 30], SOSTDC1 [31, 32], TGFβ2 [33, 34], TMEM79 [35], BMP7 [36, 37], WNT10A [38], ZDHHC21 [39], SOX10 [40, 41], ITGB4 [42], KRT14 [43], and ITGA6 [44], which are associated with the development of the epidermis and HF. They are also related to the development of the epithelium. Functional enrichment analysis showed that the DEGs were significantly enriched in negative regulation of the canonical Wnt signaling pathway, HF development, negative regulation of the BMP signaling pathway, establishment of the skin barrier, and positive regulation of epidermal cell differentiation and skin development, highlighting the central roles of these DEGs in hair morphogenesis. Notably, the fate of HFs is affected by typical Wnt/β-catenin signaling, BMP signaling, the TGF-β signaling pathway, the PI3K-Akt signaling pathway, and the Hippo signaling pathway.
Embryonic HF development and postnatal hair growth rely on intercellular communication within the epithelium and between epithelial and mesenchymal cells [50]. A homozygous missense mutation within KRT25 that causes autosomal recessive woolly hair in humans, which is consistent with findings in mutant mice. The identification of a KRT25 mutation as a cause of woolly hair in humans [51, 52]. All-trans-retinoic acid could inhibit hair follicle growth via inhibiting proliferation and inducing apoptosis of DPCs partially through the TGFβ2/Smad2/3 pathway [33]. Reddy et al. [24] found that expression of FZD1 in the placode correlates with expression of WNT10A and WNT10B in the placode. They suggest that canonical WNT signaling is likely activated in the placodes and DC of develo** hair follicles by WNT10A and WNT10B, expressed in and secreted from epithelial cells, binding to FZD1, expressed in the placode epithelium and DC. Expression of FZD1 is detected in the DP and Mx of anagen follicles, and could potentially interact with DP and Mx cells, in particular WNT10A. In postnatal HFs in full anagen, FZD1 express in the ORS. At the germ and bulbous peg stages, WNT10A and WNT10B are expressed continuously in follicular epithelium during these later stages of morphogenesis. At the bulbous peg stage, WNT10A and WNT10B are strongly expressed in a cone of epithelial cells surrounding the DP. In rat whisker HFs WNT10A was expressed in the ORS, IRS, Mx and HS of anagen follicles [53].

a Diagrams of HF development in Merino sheep. b Correspondence between different cell types and DEGs. Epi epidermal, Pc placode, DC dermal condensate, DP dermal papilla, Mx matrix, SG sebaceous glands, SwG Sweat gland, AMP Arrector pili muscle, Mc melanocytes, ORS outer root sheath, IRS inner root sheath, PF primary follicle, SF secondary follicle, SD secondary-derived follicle, HS hair shaft
The characteristics of HF development and postpartum regeneration are significantly altered in microanatomy and cell viability experiments. HF development is controlled by a variety of signaling pathways, transcription factors and epigenetic regulatory factors (including miRNAs) [54]. Some studies based on HF gene regulatory networks have shown that the Wnt [10], TGF-β [55, 56], MAPK [57], Shh [58], BMP [59], PI3K-Akt [77, 78] is determined by controlling FDR (false discovery rate), and the corrected P-value is Q-value. At the same time, we calculated the differential expression multiple (fold change) according to the FPKM value. The screening conditions of differential genes are as follows: Q-value ≤ 0.05 and fold change ≥ 2 were considered differentially expressed between the adjacent comparison groups (comparisons: G2/G1, G3/G2 G4/G3, G5/G4, G6/G5, and G6/G1) [79]. The expression patterns of DEGs were analyzed by systematic clustering to explore the similarities and relationships between the different libraries. Furthermore, the DEGs were subjected to K-means clustering using the Euclidean distance method associated with complete linkage on the BMK Cloud platform (https://www.biocloud.net/).
Functional enrichment analysis and gene annotation
Functional annotation was performed using the GO (http://geneontology.org) and KEGG (http://www.genome.ad.jp/kegg/) databases [80] based on GO and KEGG pathways. These analyses were conducted for the DEGs identified in each tissue at each stage of HF development, which were significantly enriched in dynamic expression patterns. The BPs and metabolic pathways significantly associated with the gene lists were determined based on their FDR [81]. Functional evidence was obtained on the basis of the relationships between the significant GO terms (FDR < 0.05) and the DEGs [82]. The Database for Annotation Visualization and Integrated Discovery (DAVID) 6.8 (https://david.ncifcrf.gov/tools.jsp) was used to perform functional annotation of the genes that were significantly enriched in the expression patterns [83].
Construction of a PPI network
Following the integration of the protein information in the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) (https://string-db.org/) database with the DEGs, a PPI network of the identified DEGs was established. PPI network analysis was performed using STRING and Cytoscape software V3.8.0 (http://www.cytoscape.org/). First, STRING was utilized to analyze the correlation coefficients between genes. Interacting pairs with confidence scores > 0.5 were selected to build the PPI network, and the Matthews correlation coefficient (MCC) algorithm was used to calculate hub genes and the selected top 120 genes were visualized using Cytoscape software.
K-means analysis and WGCNA
DEGs were clustered with the R package K-means function, where K = 10 within the cluster package according to the Euclidean distance. WGCNA [84, 85] was applied to the FPKM expression data. A coexpression network was constructed with a beta value of 4. We calculated the coefficients of gene dissimilarity, performed hierarchical clustering of the genes and then determined the gene modules by the dynamic tree cut method. Through clustering analysis, modules close to each other were merged into new modules. Before WGCNA, we identified and filtered the selected gene set. We removed low-quality genes with an unstable impact on the results to improve the accuracy of network construction. The filtering criteria in this study were as follows: for each gene, the maximum count value in all samples was > 50, and the count was > 20 in at least 16 samples. The modules were functionally annotated using DAVID. Highly connected genes in each module, which are also known as hub genes, may play important roles in the module. Hub genes are conserved to a certain extent and are at the core of the gene coexpression network. These genes can act as genetic buffers to reduce the impacts of other gene mutations. We identified the top 150 hub genes in the modules that were most closely related to HF development differences, that is, the 150 genes with the highest connectivity in the modules, and used Cytoscape software to map the gene–gene interaction network to visualize the gene relationships.
Validation of RNA-seq data
Several differentially expressed mRNAs involved in HF development were selected and confirmed by RT–PCR with GAPDH as an internal reference. The primers used for RT–PCR are listed in Additional file 11: Table S7. Total RNA from the samples used for high-throughput RNA-seq was isolated and converted into cDNA using a PrimeScript™ RT Reagent Kit with gDNA Eraser (TaKaRa, Japan). RT–PCR was carried out on a CFX96™ Real-Time System (Bio–Rad, USA) using a TB Green Premix Ex Taq™ kit (TaKaRa, Japan) according to the manufacturer’s instructions. The thermal cycling conditions used in RT–PCR were 95 °C for 30 s followed by 40 cycles of 95 °C for 5 s and 60 °C for 30 s. A reaction volume of 20 μL was used for RT–PCR according to the manufacturer’s protocol. The specificity of the SYBR Green PCR signal was confirmed by melting curve analysis. The RT–PCR experiments were performed in triplicate, and the average Ct value was used for further analysis. The 2−ΔΔCt method was used to determine the relative mRNA abundance.
Availability of data and materials
All RNA-seq data generated in this study were submitted to the NCBI SRA database under BioProject No. PRJNA705554.
Abbreviations
- HF:
-
Hair follicle
- DEGs:
-
Differentially expressed genes
- SBM:
-
Subo merino
- PF:
-
Primary follicle
- SF:
-
Secondary follicle
- SD:
-
Secondary-derived follicle
- Pc:
-
Placode; DC: Dermal condensate
- DP:
-
Dermal papilla
- SG:
-
Sebaceous gland
- ORS:
-
Outer root sheath
- IRS:
-
Inner root sheath
- DF:
-
Dermal fibroblast
- Mx:
-
Matrix
- Mc:
-
Melanocytes
- Epi:
-
Epidermal
- SwG:
-
Sweat gland
- HS:
-
Hair shaft
- AMP:
-
Arrector pili muscle
- BMP:
-
Bone morphogenetic protein
- TGF-β:
-
Transforming growth factor beta
- GO:
-
Gene ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- BP:
-
Biological processes
- CAM:
-
Cell adhesion molecule
- HCM:
-
Hypertrophic cardiomyopathy
- ECM:
-
Extracellular matrix
- PPAR:
-
Peroxisome proliferator-activated receptor
- MAPK:
-
Mitogen-activated protein kinase
- mRNA:
-
Messenger RNA
- NB:
-
Newborn
- PBS:
-
Phosphate-buffered saline
- RIN:
-
RNA integrity number
- FPKM:
-
Fragments per kilobase of exon model per million mapped fragments
- FC:
-
Fold change
- FDR:
-
False discovery rates
- DAVID:
-
Database for Annotation, Visualization, and Integrated Discovery
- WGCNA:
-
Weighted gene co-expression network analysis
- PPI:
-
Protein–protein interaction
- RT–PCR:
-
Real-time PCR
- STRING:
-
Search Tool for the Retrieval of Interacting Genes/Proteins
References
Sulayman A, Tian K, Huang X, Tian Y, Xu X, Fu X, et al. Genome-Wide Identification and Characterization of Long Non-Coding RNAs Expressed During Sheep Fetal and Postnatal Hair Follicle Development. Sci Rep. 2019;9(1):8501. https://doi.org/10.1038/s41598-019-44600-w.
Galbraith H. Fundamental Hair Follicle Biology and Fine Fibre Production in Animals. Animal. 2010;4(9):1490–509. https://doi.org/10.1017/S175173111000025X.
Hardy H, Lyne A. The Pre-Natal Development of Wool Follicles in Merino Sheep. Aust J Biol Sci. 1956;9(3):421–41.
Brook AH, Short BF, Lyne AG. Formation of New Wool Follicles in the Adult Sheep. Nature. 1960;185:51. https://doi.org/10.1038/185051a0.
Chapman RE, Hopkins PS, Thorburn GD. The Effects of Fetal Thyroidectomy and Thyroxine Administration on the Development of Skin and Wool Follicles of Sheep Fetuses. J Anat. 1974; 117(Pt 2):419–32. https://www.ncbi.nlm.nih.gov /pmc/articles/PMC1231415/pdf/janat00385–0188.
Hutchison G, Mellor DJ. Effects of Maternal Nutrition on the Initiation of Secondary Wool Follicles in Foetal Sheep. J Comp Pathol. 1983;93(4):577–83. https://doi.org/10.1016/0021-9975(83)90064-6.
Chapman RE, Hardy MH. Effects of Intradermally Injected and Topically Applied Mouse Epidermal Growth Factor on Wool Growth, Skin and Wool Follicles of Merino Sheep. Aust J Biol Sci. 1988;41(2):261–8. https://doi.org/10.1071/bi9880261.
Hocking Edwards JE. Reduction in Wool Follicles Prior to Birth in Merino Sheep. Reprod Fert Develop. 1999;11(4–5):229–34. https://doi.org/10.1071/rd99049.
du Cros DL, Isaacs K, Moore GP. Localization of Epidermal Growth Factor Immunoreactivity in Sheep Skin During Wool Follicle Development. J Invest Dermatol. 1992;98(1):109–15. https://doi.org/10.1111/1523-1747.ep12496010.
Andl T, Reddy ST, Gaddapara T, Millar SE. WNT Signals are Required for the Initiation of Hair Follicle Development. Dev Cell. 2002;2(5):643–53. https://doi.org/10.1016/S1534-5807(02)00167-3.
Foitzik K, Lindner G, Mueller Roever S, Maurer M, Botchkareva N, Botchkarev V, et al. Control of Murine Hair Follicle Regression (Catagen) by TGF-β1 in Vivo. FASEB J. 2000;14(5):752–60. https://doi.org/10.1096/fasebj.14.5.752.
Plikus MV, Mayer JA, de la Cruz D, Baker RE, Maini PK, Maxson R, Chuong C. Cyclic Dermal BMP Signalling Regulates Stem Cell Activation During Hair Regeneration. Nature. 2008;451(7176):340–4. https://doi.org/10.1038/nature06457.
Sander G, Simon Bawden C, Hynd PI, Nesci A, Rogers G, Powell BC. Expression of the Homeobox Gene, Barx2, Wool Follicle Development. J Invest Dermatol. 2000;115(4):753–6. https://doi.org/10.1046/j.1523-1747.2000.00122.x.
Rogers GE. Biology of the Wool Follicle: An Excursion into a Unique Tissue Interaction System Waiting to be Re-Discovered. Exp Dermatol. 2006;15(12):931–49. https://doi.org/10.1111/j.1600-0625.2006.00512.x.
Zhao B, Fu X, Tian K, Huang X, Di J, Bai Y, et al. Identification of SNPs and Expression Patterns of FZD3 Gene and its Effect on Wool Traits in Chinese Merino Sheep (**njiang Type). J Integr Agr. 2019;18(10):2351–60. https://doi.org/10.1016/S2095-3119(19)62735-8.
Valerio C, Marianna A, Roberta E, Alfredo C. RNA-Seq and Human Complex Diseases: Recent Accomplishments and Future Perspectives. Eur J Hum Genet. 2013;21(2):134–42. https://doi.org/10.1038/ejhg.2012.129.
Zhao B, Luo H, He J, Huang X, Chen S, Fu X, et al. Comprehensive Transcriptome and Methylome Analysis Delineates the Biological Basis of Hair Follicle Development and Wool-Related Traits in Merino Sheep. BMC Biol. 2021;19(1):197. https://doi.org/10.1186/s12915-021-01127-9.
Liu N, Li H, Liu K, Yu J, Bu R, Cheng M, et al. Identification of Skin-Expressed Genes Possibly Associated with Wool Growth Regulation of Aohan Fine Wool Sheep. BMC Genet. 2014;15(1):144. https://doi.org/10.1186/s12863-014-0144-1.
Rile N, Liu Z, Gao L, Qi J, Zhao M, **e Y, et al. Expression of Vimentin in Hair Follicle Growth Cycle of Inner Mongolian Cashmere Goats. BMC Genomics. 2018;19(1):38. https://doi.org/10.1186/s12864-017-4418-7.
Liu Y, Wang L, Li X, Han W, Yang K, Wang H, et al. High-Throughput Sequencing of Hair Follicle Development-Related MicroRNAs in Cashmere Goat at Various Fetal Periods. Saudi J Biol Sci. 2018;25(7):1494–508. https://doi.org/10.1016/j.sjbs.2017.12.009.
Gao Y, Wang X, Yan H, Zeng J, Ma S, Niu Y, et al. Comparative Transcriptome Analysis of Fetal Skin Reveals Key Genes Related to Hair Follicle Morphogenesis in Cashmere Goats. PLoS ONE. 2016;11(3): e151118. https://doi.org/10.1371/journal.pone.0151118.
Danilenko DM, Ring BD, Yanagihara D, Benson W, Wiemann B, Starnes CO, Pierce GF. Keratinocyte Growth Factor is an Important Endogenous Mediator of Hair Follicle Growth, Development, and Differentiation. Normalization of the Nu/Nu Follicular Differentiation Defect and Amelioration of Chemotherapy-Induced Alopecia. Am J Pathol. 1995;147(1):145–54.
Yang L, Yamasaki K, Shirakata Y, Dai X, Tokumaru S, Yahata Y, et al. Bone Morphogenetic Protein-2 Modulates Wnt and Frizzled Expression and Enhances the Canonical Pathway of Wnt Signaling in Normal Keratinocytes. J Dermatol Sci. 2006;42(2):111–9. https://doi.org/10.1016/j.jdermsci.2005.12.011.
Reddy ST, Andl T, Lu MM, Morrisey EE, Millar SE. Expression of Frizzled Genes in Develo** and Postnatal Hair Follicles. J Invest Dermatol. 2004;123(2):275–82. https://doi.org/10.1111/j.0022-202X.2004.23215.x.
Zhang C, Li Y, Qin J, Yu C, Ma G, Chen H, Xu X. TMT-Based Quantitative Proteomic Analysis Reveals the Effect of Bone Marrow Derived Mesenchymal Stem Cell on Hair Follicle Regeneration. Front Pharmacol. 2021; 12. https://doi.org/10.3389/fphar.2021.658040.
Morgan HJ, Benketah A, Olivero C, Rees E, Ziaj S, Mukhtar A, et al. Hair Follicle Differentiation-Specific Keratin Expression in Human Basal Cell Carcinoma. Clin Exp Dermatol. 2020;45(4):417–25. https://doi.org/10.1111/ced.14113.
Li J, Tzu J, Chen Y, Zhang YP, Nguyen NT, Gao J, et al. Laminin-10 is Crucial for Hair Morphogenesis. Embo J. 2003;22(10):2400–10. https://doi.org/10.1093/emboj/cdg239.
Wegner J, Loser K, Apsite G, Nischt R, Eckes B, Krieg T, et al. Laminin α5 in the Keratinocyte Basement Membrane is Required for Epidermal-Dermal Intercommunication. Matrix Biol. 2016;56:24–41. https://doi.org/10.1016/j.matbio.2016.05.001.
Ahn Y, Sims C, Logue JM, Weatherbee SD, Krumlauf R. Lrp4 and Wise Interplay Controls the Formation and Patterning of Mammary and Other Skin Appendage Placodes by Modulating Wnt Signaling. Development (Cambridge). 2013;140(3):583–93. https://doi.org/10.1242/dev.085118.
Ohazama A, Johnson EB, Ota MS, Choi HJ, Porntaveetus T, Oommen S, et al. Lrp4 Modulates Extracellular Integration of Cell Signaling Pathways in Development. PLoS ONE. 2008;3(12): e4092. https://doi.org/10.1371/journal.pone.0004092.
Arikan V, Cumaogullari O, Ozgul BM, Oz FT. Investigation of SOSTDC1 Gene in Non-Syndromic Patients with Supernumerary Teeth. Med Oral Patol Oral Cir Bucal. 2018:0–0. https://doi.org/10.4317/medoral.22520.
Närhi K, Tummers M, Ahtiainen L, Itoh N, Thesleff I, Mikkola ML. Sostdc1 Defines the Size and Number of Skin Appendage Placodes. Dev Biol. 2012;364(2):149–61. https://doi.org/10.1016/j.ydbio.2012.01.026.
Nan W, Li G, Si H, Lou Y, Wang D, Guo R, Zhang H. All-Trans-Retinoic Acid Inhibits Mink Hair Follicle Growth Via Inhibiting Proliferation and Inducing Apoptosis of Dermal Papilla Cells through TGF-β2/Smad2/3 Pathway. Acta Histochem. 2020;122(7): 151603. https://doi.org/10.1016/j.acthis.2020.151603.
Kim B, Yoon SK. Hairless Up-Regulates Tgf-β2 Expression via Down-Regulation of miR-31 in the Skin of “Hairpoor” (HrHp) Mice. J Cell Physiol. 2015;230(9):2075–85. https://doi.org/10.1002/jcp.24935.
Elias PM, Wakefield JS. Mechanisms of Abnormal Lamellar Body Secretion and the Dysfunctional Skin Barrier in Patients with Atopic Dermatitis. J Allergy Clin Immunol. 2014;134(4):781–91. https://doi.org/10.1016/j.jaci.2014.05.048.
Lv X, Gao W, ** C, Wang L, Wang Y, Chen W, et al. Preliminary Study on microR-148a and microR-10a in Dermal Papilla Cells of Hu Sheep. BMC Genet. 2019;20(1):70. https://doi.org/10.1186/s12863-019-0770-8.
Kowtharapu B, Prakasam R, Murín R, Koczan D, Stahnke T, Wree A, et al. Role of Bone Morphogenetic Protein 7 (BMP7) in the Modulation of Corneal Stromal and Epithelial Cell Functions. Int J Mol Sci. 2018;19(5):1415. https://doi.org/10.3390/ijms19051415.
Wang K, Yamada S, Izumi H, Tsukamoto M, Nakashima T, Tasaki T, et al. Critical in Vivo Roles of WNT10A in Wound Healing by Regulating Collagen Expression/Synthesis in WNT10A-deficient Mice. PLoS ONE. 2018;13(3): e195156. https://doi.org/10.1371/journal.pone.0195156.
Mill P, Lee AW, Fukata Y, Tsutsumi R, Fukata M, Keighren M, et al. Palmitoylation Regulates Epidermal Homeostasis and Hair Follicle Differentiation. PloS Genet. 2009;5(11): e1000748. https://doi.org/10.1371/journal.pgen.1000748.
Lezcano C, Ho J, Seethala RR. Sox10 and DOG1 Expression in Primary Adnexal Tumors of the Skin. Am J Dermatopathol. 2017;39(12):896–902. https://doi.org/10.1097/DAD.0000000000000872.
Slater NA, Googe PB. Sox10 Positive Breast Carcinoma Metastatic to the Skin. J Cutan Pathol. 2018;45(5):373–4. https://doi.org/10.1111/cup.13114.
Wee LWY, Tan EC, Bishnoi P, Ng YZ, Lunny DP, Lim HW, et al. Epidermolysis Bullosa with Pyloric Atresia Associated with Compound Heterozygous ITGB4 Pathogenic Variants: Minimal Skin Involvement but Severe Mucocutaneous Disease. Pediatr Dermatol. 2021;38(4):908–12. https://doi.org/10.1111/pde.14668.
Gunnarsson AP, Christensen R, Li J, Jensen UB. Dataset on Gene Expression Profiling of Multiple Murine Hair Follicle Populations. Data Brief. 2016;9:328–34. https://doi.org/10.1016/j.dib.2016.08.063.
Chen Z, Shen G, Tan X, Qu L, Zhang C, Ma L, et al. ID1/ID3 Mediate the Contribution of Skin Fibroblasts to Local Nerve Regeneration through Itga6 in Wound Repair. Stem Cell Transl Med. 2021;10(12):1637–49. https://doi.org/10.1002/sctm.21-0093.
Rendl M, Lewis L, Fuchs E. Molecular Dissection of Mesenchymal-Epithelial Interactions in the Hair Follicle. PloS Biol. 2005;3(11): e331. https://doi.org/10.1371/journal.pbio.0030331.
Yang F, Li R, Zhao C, Che T, Guo J, **e Y, et al. Single-Cell Sequencing Reveals the New Existence form of Dermal Papilla Cells in the Hair Follicle Regeneration of Cashmere Goats. Genomics. 2022;114(2): 110316. https://doi.org/10.1016/j.ygeno.2022.110316.
Ge W, Tan SJ, Wang SH, Li L, Sun XF, Shen W, Wang X. Single-Cell Transcriptome Profiling Reveals Dermal and Epithelial Cell Fate Decisions During Embryonic Hair Follicle Development. Theranostics. 2020;10(17):7581–98. https://doi.org/10.7150/thno.44306.
Wang S, Wu T, Sun J, Li Y, Yuan Z, Sun W. Single-Cell Transcriptomics Reveals the Molecular Anatomy of Sheep Hair Follicle Heterogeneity and Wool Curvature. Front Cell Dev Biol. 2021; 9. https://doi.org/10.3389/fcell.2021.800157.
Amirfakhryan E, Davarnia B, Jeddi F, Najafzadeh N. Azelaic Acid Stimulates Catalase Activation and Promotes Hair Growth through Upregulation of Gli1 and Gli2 mRNA and Shh Protein. Avicenna J Phytomed. 2020; 10(5):460–71. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7508322/pdf/AJP-10-460.
Lintern KB, Guidato S, Rowe A, Saldanha JW, Itasaki N. Characterization of Wise Protein and its Molecular Mechanism to Interact with Both Wnt and BMP Signals. J Biol Chem. 2009;284(34):23159–68. https://doi.org/10.1074/jbc.M109.025478.
Zernov NV, Skoblov MY, Marakhonov AV, Shimomura Y, Vasilyeva TA, Konovalov FA, et al. Autosomal Recessive Hypotrichosis with Woolly Hair Caused by a Mutation in the Keratin 25 Gene Expressed in Hair Follicles. J Invest Dermatol. 2016;136(6):1097–105. https://doi.org/10.1016/j.jid.2016.01.037.
Yu X, Chen F, Ni C, Zhang G, Zheng L, Zhang J, et al. A Missense Mutation within the Helix Termination Motif of KRT25 Causes Autosomal Dominant Woolly Hair/Hypotrichosis. J Invest Dermatol. 2018;138(1):230–3. https://doi.org/10.1016/j.jid.2017.08.035.
Lin C, Yuan Y, Chen X, Li H, Cai B, Liu Y, et al. Expression of Wnt/β-catenin Signaling, Stem-Cell Markers and Proliferating Cell Markers in Rat Whisker Hair Follicles. J Mol Histol. 2015;46(3):233–40. https://doi.org/10.1007/s10735-015-9616-5.
Andl T, Botchkareva NV. MicroRNAs (miRNAs) in the Control of HF Development and Cycling: The Next Frontiers in Hair Research. Exp Dermatol. 2016;24(11):821–6. https://doi.org/10.1111/exd.12785.
Foitzik K, Paus R, Doetschman T, Dotto GP. The TGF-β2 Isoform is Both a Required and Sufficient Inducer of Murine Hair Follicle Morphogenesis. Dev Biol. 1999;212(2):278–89. https://doi.org/10.1006/dbio.1999.9325.
Massagué J. How Cells Read TGF-β Signals. Nat Rev Mol Cell Bio. 2000;1(3):169–78. https://doi.org/10.1038/35043051.
Headon DJ, Overbeek PA. Involvement of a Novel Tnf Receptor Homologue in Hair Follicle Induction. Nat Genet. 1999;22(4):370–4. https://doi.org/10.1038/11943.
Kata B, Hamel PA. Alx4 Binding to LEF-1 Regulates N-CAM Promoter Activity. J Biol Chem. 2002;277(2):1120–7. https://doi.org/10.1074/jbc.M109912200.
Botchkarev VA, Sharov AA. BMP Signaling in the Control of Skin Development and Hair Follicle Growth. Differentiation. 2010;72(9–10):512–26. https://doi.org/10.1111/j.1432-0436.2004.07209005.x.
Cai B, Zheng Y, Ma S, **ng Q, Wang X, Yang B, et al. Long Non-Coding RNA Regulates Hair Follicle Stem Cell Proliferation and Differentiation through PI3K/AKT Signal Pathway. Mol Med Rep. 2018;17(4):5477–83. https://doi.org/10.3892/mmr.2018.8546.
Chen Y, Fan Z, Wang X, Mo M, Zeng SB, Xu RH, et al. PI3K/Akt Signaling Pathway is Essential for De Novo Hair Follicle Regeneration. Stem Cell Res Ther. 2020;11(1):144. https://doi.org/10.1186/s13287-020-01650-6.
Serrano CH, Ospina JP, Salazar PL, Cardona-Castro N. Notch Signaling Pathway Expression in the Skin of Leprosy Patients: Association with Skin and Neural Damage. Front Immunol. 2020; 11. https://doi.org/10.3389/fimmu.2020.00368.
Efrat AK, Torres IL, Schejter ED, Daniel St J, Ben-Zion S. Drosophila Follicle Cells are Patterned by Multiple Levels of Notch Signaling and Antagonism Between the Notch and JAK/STAT Pathways. Development. 2007;134(6):1161–9. https://doi.org/10.1242/dev.02800.
Gentile P, Garcovich S. Advances in Regenerative Stem Cell Therapy in Androgenic Alopecia and Hair Loss: Wnt pathway, Growth-Factor, and Mesenchymal Stem Cell Signaling Impact Analysis on Cell Growth and Hair Follicle Development. Cells-Basel. 2019;8(5):466. https://doi.org/10.3390/cells8050466.
Paus R, Foitzik K, Welker P, Bulfone-Paus S, Eichmuller S. Transforming Growth Factor-Beta Receptor Type I and Type II Expression During Murine Hair Follicle Development and Cycling. J Invest Dermatol. 1997;109(4):518–26. https://doi.org/10.1111/1523-1747.ep12336635.
Luo K. Signaling Cross Talk Between TGF-β/Smad and Other Signaling Pathways. Csh Perspect Biol. 2017;9(1): a22137. https://doi.org/10.1101/cshperspect.a022137.
Plikus MV, Baker RE, Chih-Chiang C, Clyde F, Damon DLC, Thomas A, et al. Self-Organizing and Stochastic Behaviors During the Regeneration of Hair Stem Cells. Science. 2011;332(6029):586. https://doi.org/10.1126/science.1201647.
Botchkarev VA, Yaar M, Peters EMJ, Raychaudhuri SP, Botchkareva NV, Marconi A, et al. Neurotrophins in Skin Biology and Pathology. J Invest Dermatol. 2006;126(8):1719–27. https://doi.org/10.1038/sj.jid.5700270.
Kamberov YG, Karlsson EK, Kamberova GL, Lieberman DE, Sabeti PC, Morgan BA, Tabin CJ. A Genetic Basis of Variation in Eccrine Sweat Gland and Hair Follicle Density. P Natl Acad Sci Usa. 2015;112(32):9932. https://doi.org/10.1073/pnas.1511680112.
Thiboutot D. Regulation of Human Sebaceous Glands. J Invest Dermatol. 2004;123(1):1–12. https://doi.org/10.1111/j.1523-1747.2004.t01-2-.x.
Smith KR, Thiboutot DM. Thematic Review Series: Skin Lipids. Sebaceous Gland Lipids: Friend or Foe? J Lipid Res. 2008;49(2):271–81.
Hanley K, Jiang Y, Crumrine D, Bass NM, Appel R, Elias PM, et al. Activators of the Nuclear Hormone Receptors PPAR alpha and FXR Accelerate the Development of the Fetal Epidermal Permeability Barrier. J Clin Invest. 1997;100(3):705–12. https://doi.org/10.1172/JCI119583.
Hanley K, Kömüves LG, Bass NM, He SS, Yan J, Crumrine D, et al. Fetal Epidermal Differentiation and Barrier Development in Vivo is Accelerated by Nuclear Hormone Receptor Activators 1. J Invest Dermatol. 1999;113(5):788–95. https://doi.org/10.1046/j.1523-1747.2000.00073.x.
Tian Y, Yang X, Du J, Zeng W, Wu W, Di J, et al. Differential Methylation and Transcriptome Integration Analysis Identified Differential Methylation Annotation Genes and Functional Research Related to Hair Follicle Development in Sheep. Front Genet. 2021; 12. https://doi.org/10.3389/fgene.2021.735827.
Trapnell C, Pachter L, Salzberg SL. TopHat: Discovering Splice Junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–11. https://doi.org/10.1093/bioinformatics/btp120.
Robinson MD, McCarthy DJ, Smyth GK. EdgeR: A Bioconductor Package for Differential Expression Analysis of Digital Gene Expression Data. Bioinformatics. 2009;26(1):139–40. https://doi.org/10.1093/bioinformatics/btp616.
Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological). 1995; 57(1). https://doi.org/10.2307/2346101.
Benjamini Y, Yekutieli D. The Control of the False Discovery Rate in Multiple Testing Under Dependency. Ann. Statist. 2001; 29(4). https://doi.org/10.1214/aos/1013699998.
Ren H, Wang G, Chen L, Jiang J, Liu L, Li N, et al. Genome-Wide Analysis of Long Non-Coding RNAs at Early Stage of Skin Pigmentation in Goats (Capra Hircus). BMC Genomics. 2016; 17(1). https://doi.org/10.1186/s12864-016-2365-3.
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. https://doi.org/10.1093/nar/28.1.27.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: Tool for the Unification of Biology. The Gene Ontology Consortium Nat Genet. 2000;25(1):25–9. https://doi.org/10.1038/75556.
Cardoso TF, Cánovas A, Canela XO, González PR, Amills M, Quintanilla R. RNA-seq Based Detection of Differentially Expressed Genes in the Skeletal Muscle of Duroc Pigs with Distinct Lipid Profiles. Sci Rep-Uk. 2017; 7(1). https://doi.org/10.1038/srep40005.
Cánovas A, Pena RN, Gallardo D, Ramírez O, Amills M, Quintanilla R, Moore S. Segregation of Regulatory Polymorphisms with Effects on the Gluteus Medius Transcriptome in a Purebred Pig Population. PLoS ONE. 2012;7(4): e35583. https://doi.org/10.1371/journal.pone.0035583.
Langfelder P, Horvath S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinformatics. 2008;9(1):559. https://doi.org/10.1186/1471-2105-9-559.
Wu Z, Hai E, Di Z, Ma R, Shang F, Wang Y, et al. Using WGCNA (Weighted Gene Co-Expression Network Analysis) to Identify the Hub Genes of Skin Hair Follicle Development in Fetus Stage of Inner Mongolia Cashmere Goat. PLoS ONE. 2020;15(12): e243507. https://doi.org/10.1371/journal.pone.0243507.
Acknowledgements
The authors gratefully acknowledge the constructive comments from three reviewers
Funding
This work was supported by the China Agriculture Research System (Nos. CARS-39) and Agricultural science and technology innovation project of Shandong Academy of Agricultural Sciences (Nos. CXGC2021B20). The funders played no role in study design, collection, analysis, data interpretation, manuscript writing, or decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
JH and KT conceived and designed the study. JH, BZ, XH, and GL collected the samples and/or generated the data. JH, CW, JM, and JL conducted the statistical analyses. XF and YT managed the experimental sheep populations. JH, BZ, SG and KT drafted and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was conducted in accordance with the Basel Declaration and it is reported in accordance with ARRIVE guidelines. Collection of all tissue samples was conducted in accordance with the guidelines of the Ethics Committee of the College of Animal Science (approval number: 2006–398) of Gansu Agricultural University. All embryo samples were obtained by cesarean section after 12 ewes were electrocution followed by exsanguination. The skin tissues were collected immediately after euthanasia, used for scientific research. On postnatal days P7 and P30, lamb was observed to lose consciousness after using general anesthetic the skin tissue of postnatal lambs was collected in vivo, with a depth of about 2 cm2 × 3 mm. Then skin wounds were quickly sprinkled anti-inflammatory powder and carefully suture. We took good care of the lamb until the wound healed. All efforts were made to minimize animal suffering and discomfort.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Result Statistics of mRNA Genome Comparison. Map** ratio=Mapped reads/All reads, Mapped Unique reads: reads that have only one position in the genome.
Additional file 2:
Fig. S1. Heatmap of DEGs during the hair follicle morphogenesis. The x-axis represents the sample number, where S1 to S3 is G1; S4 to S6 is G2; S7 to S9 is G3; S10 to S12 is G4; S13 to S15 is G5; S16 to S18 is G6.
Additional file 3:
Fig. S2. Enrichment analysis of all DEGs. (a)Top 30 of GO enrichment; (b)Top 30 of pathway enrichment.
Additional file 4:
Table S2. GO enrichment of DEGs.
Additional file 5:
Table S3. KEGG pathway of DEGs.
Additional file 6: Fig. S3.
Go and KEGG network diagram during the hair follicle morphogenesis. (a-f) represents the comparison group of G1-G6. Circle is represented gene, square is represented KEGG; triangle is represented GO term.
Additional file 7: Fig. S4.
K-means clustering analysis of differentially expressed mRNAs among the six comparison groups.
Additional file 8:
Table S4. GO analysis of K-means clusters.
Additional file 9:
Table S5. GO analysis of module in WGCNA.
Additional file 10:
Table S6. KEGG pathway of module in WGCNA.
Additional file 11:
Table S7. Sequence of primers.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
He, J., Zhao, B., Huang, X. et al. Gene network analysis reveals candidate genes related with the hair follicle development in sheep. BMC Genomics 23, 428 (2022). https://doi.org/10.1186/s12864-022-08552-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-022-08552-2