
Abstract
Background
Apparent Amylose Content (AAC), regulated by the Waxy gene, represents the key determinant of rice cooking properties. In occidental countries high AAC rice represents the most requested market class but the availability of molecular markers allowing specific selection of high AAC varieties is limited.
Results
In this study, the effectiveness of available molecular markers in predicting AAC was evaluated in a collection of 127 rice accessions (125 japonica ssp. and 2 indica ssp.) characterized by AAC values from glutinous to 26%. The analyses highlighted the presence of several different allelic patterns identifiable by a few molecular markers, and two of them, i.e., the SNPs at intron1 and exon 6, were able to explain a maximum of 79.5% of AAC variation. However, the available molecular markers haplotypes did not provide tools for predicting accessions with AAC higher than 24.5%. To identify additional polymorphisms, the re-sequencing of the Waxy gene and 1kbp of the putative upstream regulatory region was performed in 21 genotypes representing all the AAC classes identified. Several previously un-characterized SNPs were identified and four of them were used to develop dCAPS markers.
Conclusions
The addition of the SNPs newly identified slightly increased the AAC explained variation and allowed the identification of a haplotype almost unequivocally associated to AAC higher than 24.5%. Haplotypes at the waxy locus were also associated to grain length and length/width (L/W) ratio. In particular, the SNP at the first intron, which identifies the Wxa and Wxb alleles, was associated with differences in the width of the grain, the L/W ratio and the length of the kernel, most likely as a result of human selection.
Similar content being viewed by others

Background
Rice (Oryza sativa L.) is the most important staple food for over half of the world population. Rice quality is primarily influenced by starch which is composed of two polysaccharides: amylose and amylopectin. The percentage of amylose on total starch, measured as Apparent Amylose Content (AAC), is the key determinant of rice cooking properties. High amylose varieties, like risotto varieties, cook dry with firm and separate grain; while low amylose cultivars (cvs.) are tender, glossy and cohesive after cooking. Suggested classification of amylose content identified classes as waxy (0–5%), very low (5–12%), low (12–20%), intermediate (20–25%), and high (25–33%), even considering that commercially rice is classified by amylose content as either low (less than 20% amylose), medium (21–25%) and high (26–33%) (Juliano 1992; Suwannaporn et al. 2007).
Over the past several decades, various methods have been reported for the determination of amylose content, including iodine binding, near infrared spectroscopy, size-exclusion chromatography and most recently, asymmetric field flow fractionation (Juliano 1971; Wesley et al. 2003; Ward et al. 2006; Chiaramonte et al. 2012). However, none of these methods is cost-effective in terms of high throughput screening (Caffagni et al. 2013) and the iodine binding represents the only one which has been validated for routine use (Fitzgerald et al. 2008). The Waxy gene is located on chromosome 6 and consists of 13 exons and 12 introns. Two wild type alleles, Wxa, primarily found in indica subspecies, and Wxb, mainly found in japonica subspecies, have been found to predominate at the waxy locus for high and low AAC respectively (Dobo et al. 2010). The difference between the two alleles is related to the presence in Wxb of a G to T Single Nucleotide Polymorphism (SNP) at the 5′ splice site of the first 1,124 bp long intron, localized 1,164 bp upstream the start codon (haplotype AGT T instead of AGG T). Most of the waxy and low AAC cvs. screened so far carry this polymorphism which results, for Wxb, in the reduction of pre-mRNA splicing efficiency and promotion of alternative splicing at cryptic sites in exon 1, leading to a decreased production of functional enzymes and causing the glutinous and low amylose phenotypes (Wang et al. 1995; Ayres et al. 1997; Bligh et al. 1998; Cai et al. 2006). Moreover, Takanokai and co-workers (2009) compared the sequence of the GS3 gene, responsible of grain size, in 54 rice cvs. and identified 86 SNPs and 20 InDels. The allele mining for the rice fragrant gene badh2 also allowed the development of diagnostic molecular markers (Shi et al. 2008).
Allele mining experiments applied to the Waxy gene led to the identification of five different allelic variants. Mikami et al. (2008) studied the allelic diversification at the Wx locus in Asian rice associating different alleles to grain amylose content alterations. They identified the Wxop allele in indica varieties from India, Nepal, Indonesia and China showing an opaque and chalky endosperm with a very low amylose content. This allele is characterized by an A to G SNP in exon 4 at position +715 from the ATG causing an Asp to Gly aminoacid change. The allele Wxin was frequent in accessions belonging to an aromatic group and to tropical japonica which exhibited an intermediate AAC and is determined by a non-conservative mutation in exon 6 at position +1,083. The mutation alters a Tyr to Ser in the active site of the enzyme reducing its specific activity (Dobo et al. 2010). The last allele analyzed by Mikami and co-workers (2008), the wx allele, is present only in waxy varieties and is characterized by a 23 bp duplication in the second exon, 100 bp downstream the ATG, causing a premature stop codon which inactivates the Waxy gene. A minor allele, represented by Wxmq, was identified in the low AAC rice cv. Milky Queen and was characterized by two base changes within the coding region: G to A in exon 4 and T to C in exon 5. Each of these mutations generated missense aminoacid substitutions (Sato et al. 2002). One additional low AAC-associate allele, Wxhp, was identified by Liu et al. (2009) in a few low AAC Yunnan landraces. An A to G SNP occurring in exon 2 at position +497 causes an Asp to Gly substitution resulting in a reduction of the activity of GBSSI.
Following the discovery of this variability at the Wx locus, additional AAC class-specific molecular markers were developed. The combination of the Wxin SNP in exon 6 and a SNP in exon 10, which consists of a C/T non-conservative SNP, with the RM190 microsatellite in exon 1 enabled the discrimination between intermediate and high AAC in a small number of genotypes (Larkin and Park 2003). More recently, Dobo and co-workers (2010) performed the same analysis increasing the number of genotypes and identified three allelic groups based on the combination of the three SNPs explaining 89.2% of the variation in AAC among 85 US varieties and 93.8% of the variation in 279 European accessions.
To date, the attention has been focalised in the identification of Wx genetic variants explaining amylose reduction in the endosperm of intermediate, low and waxy genotypes and the molecular markers used so far can discriminate only waxy, low, intermediate and high AAC. There are no markers explaining the different percentages of seed amylose within the high amylose class and further elucidations are needed to improve rice quality particularly in occidental countries where dry and firm rice is largely preferred.
In this study a collection of 127 rice accessions (125 japonica ssp. and 2 indica ssp.) was analyzed for AAC leading to the identification of the AAC classes low, intermediate and high. Effectiveness of available markers diagnostic for Apparent Amylose Content was verified and a low predictability within the high AAC class was observed. To discover new molecular markers associated to the different AACs, twenty-one genotypes representing each AAC class were selected and subjected to the re-sequencing of the waxy locus. New SNPs were identified in four high AAC accessions and used to develop new SNP-based molecular markers. Moreover, un-expected associations between grain shape characters and polymorphisms associated to the waxy locus were identified and analyzed.
Results
AAC assessment in a rice germplasm collection
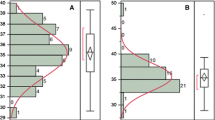
A collection of 125 temperate japonica and two indica rice accessions originated from different rice cultivation areas was evaluated for AAC and four classes were identified, ranging from waxy to high AAC (Table 1). No accessions of the very low AAC class (5–12%) were detected; high frequency was instead observed for low and intermediate amylose classes (Figure 1) while a low frequency of the high amylose class was highlighted. Accessions showing AAC higher than 20% (60 in total) originated from very different countries (20 from USA, 11 from Italy, 4 from Portugal, 3 from Spain, 2 from France and the remaining from other countries), indicating that the trait conferring a relatively high amylose content was selected in different rice cultivation areas.

Frequency distribution of amylose content classes in the analyzed germplasm collection: Waxy (0-5%), low amylose (5-20%), intermediate (20-25%) and high amylose (>25%).
Molecular markers analyses
The germplasm collection of 127 rice accessions (Table 1) was chosen to evaluate the effectiveness of known molecular markers in predicting Apparent Amylose Content. Ten different alleles for the RM190 CTn microsatellite were identified (Table 2) but a clear relationship with the different AAC groups was observed only for some alleles (Table 1). Repeats CT9, CT10 and CT14 were present only in genotypes with 23–24.85% AAC, while CT11 and CT20 also identified accessions with AAC higher than 25%. Among this AAC classes, two varieties showed unique CT alleles: CT13 for Ota (22.55% AAC) and CT21 for Bomba (22.84% AAC). The most frequent allele, CT18 identified in 67 accessions, represents a wide range of AAC (14.92-21.56%). Similar results were observed for CT17 and CT19 which were associated to heterogeneous AAC intervals ranging from 15.55% to 23.27%. Considering non-glutinous genotypes, the RM190 microsatellite explained the 74.9% of the AAC variation in our collection (Table 3).
For the SNP in the splicing site of the leader intron (intron 1), results confirming previous investigations were observed (Wang et al. 1995; Ayres et al. 1997; Bligh et al. 1998; Cai et al. 1998; Isshiki et al. 1998; Larking and Park 2003; Dobo et al. 2010). With only two exceptions represented by Rotundus and Antoni, all the accessions with AAC lower than 22% showed the T allele, while all the genotypes with AAC higher than 22% had the G allele (Table 1). This SNP explained a higher level of AAC variation (77.5%) with respect to the RM190 (Table 3) and the combination of the two polymorphisms did not significantly increase the level of explained variability (77.7%).
The presence of SNPs characterizing the different known Wx alleles was assessed in our collection through sequencing of exons 2, 4, 5 and 6. The wx allele was identified in the waxy cv. Calmochi 101, in which the 23 bp duplication (sequence motif: ACGGGTTCCAGGGCCTCAAGCCC) in exon 2 responsible for Waxy gene inactivation was present; literature data in fact provide AAC values for this cv. ranging from 0.8 to 1% (Park et al. 2007; Li et al. 2008). Allelic variation in the Wxin allele, characterized by a non-conservative A/C mutation in exon 6, was observed with the A allele being detected, with few exceptions, in accessions with AAC ranging from waxy to 22% and higher than 24%, and the C allele generally present in genotypes with an AAC from 22 to 24% (Table 1). No polymorphisms were identified in exons 4 and 5, representative of the Wxmq allele and, unlike in previous observations (Dobo et al. 2010), we did not observe mutations in exon 10 thus excluding the presence of SNPs in this exon.
Combining the results obtained for SNPs in intron 1 and exon 6, three allelic patterns were identified: GA, TA and GC (Table 2), the first letter indicating the G/T polymorphism in intron 1 while the second the A/C SNP in exon 6. Similarly to the behaviour observed for intron 1, most of the accessions from waxy to 22% AAC (with two exceptions) carried the TA haplotype (Table 1), GC pattern was present in accessions with 22-24% AAC (with two exceptions) and GA in accessions with AAC >24% (with three exceptions). Statistical analyses showed that the A/C SNP in exon 6 alone explained only the 30.9% of the variation in AAC, but altogether the two SNPs explained the 79.5%. Adding the RM190 microsatellite to the analysis, we did not find a significantly higher explanation of the AAC variation: 79.6% vs. 79.5% (Table 3).
The allelic variation at the RM190 locus together with intron 1/exon 6 SNPs data allowed the identification of allelic patterns associated to different AAC classes. In particular, even considering that no association of CT17 with a specific AAC group could be identified, all the accessions with haplotype CT17, G in intron 1 and C in exon 6 shared an AAC level ranging from 22 to 23%, with the exception of Giada (23.93%). Similarly, the CT14 allele associated with the CG and AG allelic patterns shared similar AAC (ranging from 23.2 to 23.8%). Among the CT20 group, the most frequent allelic pattern for high AAC class was CG, which identifies an AAC range from 22 to 24%, while in combination with AG frequently identified accessions with more than 25% AAC. CT9, CT10 and CT11 associated to the allelic pattern AG were typical of genotypes with more than 24% of AAC.
Despite the fact that the level of variation in AAC explained by our results is in agreement to the one recently evaluated in an Italian rice collection (Caffagni et al. 2013), it is consistently lower than previously observed. As examples, Ayres et al. (1997) using the combination of RM190 and the G/T in intron1 explained the 85.9% of the variation in AAC; Dobo and co-workers (2010) could explain the 93.8% of variation in AAC with RM190, the SNP in intron 1, the SNP in exon 6 and the SNP in exon 10 which was not present in our collection. Owing these results, it was realized that additional molecular markers could be needed to increase predictability of the different AAC classes within our germplasm panel.
Allele mining of the GBSSI gene
To mine the genetic variation at the level of the GBSSI locus within our germplasm collection, the gene as well as 1kbp of the upstream putative regulatory region were sequenced in twenty-one genotypes representing all the AAC classes identified: Calmochi 101 for the waxy type; Prever, Yrl 196, Delta, Lomellino and Campino for 14-16% AAC; Yrm 6–2, Timich 108, Augusto, Loto and Sant’Andrea for 16-19% AAC; Upla 91, Antoni, Gigante Vercelli, A201 and Gladio for 21-25% AAC; and Fragrance, L 202, Zhen Shang 47, Arroyogrande and Alinano C for >25% AAC (Table 1). The twenty-one GBSSI sequences were compared by multiple alignments considering the Nipponbare sequence as the reference. Sequence comparisons showed an overall high level of similarity (Figure 2), indicating that the coding sequence was conserved in most of the genotypes with some exceptions and led to the identification of 32 SNPs (Table 4). The waxy cv. Calmochi 101 carried the wx allele with the 23 bp duplication in exon 2, as described before. Antoni and Gigante Vercelli accessions showed the non-conservative A/C SNP in exon 6, previously identified and causing a Serine/Tyrosine substitution in the GBSSI protein (Larking and Park, 2003). Additionally, these genotypes contained two common SNPs: one in the putative regulatory region (position −1,514) and one in the first intron (position −399). Gigante Vercelli also showed a SNP at −2,174 bp upstream the ATG (Figure 2; Table 4). Zhen Shang 47 and Alinano C carried a C/T SNP at position +1,801 in exon 9 which results in the substitution of Pro415 to Ser in the GBSS protein. Also for Zhen Shang 47 and Alinano C, SNP mutations were identified in the non-coding sequences: 2 in the promoter region, 11 in intron 1, 1 in intron 6, 9 in intron 10 and 2 in intron 12 (Figure 2; Table 4). Most of the SNPs identified in the present work are not classified in the group of SNPs computationally characterized by Kharabian (2010) and present in OryzaSNP (http://oryzasnp.plantbiology.msu.edu/cgi-bin/gbrowse/osa_snp_tigr/) and dbSNP (http://www.ncbi.nlm.nih.gov/sites/entrez?db=snp&TAbCmd=Limits) databases. Additionally to these SNPs polymorphisms, a 4 bp tandem repeat that was identified in rice cvs. showing high level of Wx transcripts (Cai et al. 1998) was highlighted at position +272 in the rice accessions Alinano C and Zhen Shang 47. In these two accessions, also a 1 bp deletion was present in intron 1 at position −862 and a 3 bp deletion in intron 10 at 2,241 bp from the start codon (data not shown).

Schematic representation of sequence alignment for the GBSSI alleles. Nipponbare Wx sequence was used as reference. The position of the ATG is indicated with 0; negative numbers are referred to bp positions in the putative regulatory regions while positive numbers indicate bp positions in the coding region. Colour shade represents the different AAC classes from white corresponding to waxy to dark grey corresponding to high AAC. SNPs are symbolized by black bars, insertions by blue triangles and deletions by red crosses. Thick, intermediate and tight boxes indicate exons, 5′ and 3′ UTR and the promoter region, respectively. Lines between boxes represent introns.
Generation of new molecular markers from allele mining
With the aim of identifying more informative markers allowing a better discrimination between accessions with AAC higher than 25% from those with lower levels, one and three SNPs were selected from Antoni/Gigante Vercelli and Alinano C/Zhen Shang 47, respectively, for molecular markers development. dCAPS molecular markers were obtained from the SNPs identified at positions −1,514 (promoter region) (Antoni and Gigante Vercelli), +1,801 (exon 9), +2,282 (intron 10) and +2,806 (intron 12) (Alinano C and Zhen Shang 47) and used to screen the 127 accessions (Figure 3; Table 1). Antoni and Gigante Vercelli haplotype (T) at position −1,514 was identified in 17 additional accessions belonging to the group with AAC 20.65 – 24.85%. Among them, 11 carried the C allele for the SNP in exon 6 as Antoni and Gigante Vercelli. Considering both the SNPs, four allelic patterns associated to different levels of AAC were identified (Table 5). The AG allele (the first base referred to the SNP in exon 6, while the second to position −1,514) was present in 89 accessions and associated to a mean AAC value of 18.99% (without considering the waxy variety Calmochi 101); the GC allele was found in 18 accessions with a mean AAC value of 23.50%; the AT allele, carried by 6 accessions, corresponded to an AAC average of 24.24%; the last allele, CT, was identified in 13 genotypes with a mean AAC of 22.75%.

dCAPS molecular markers developed from SNPs found in Antoni, Gigante Vercelli, Alinano C and Zhen Shang 47 Waxy alleles. Lomellino and Prever were used as control. Primers and restriction enzymes for each molecular marker are listed in Additional file 1: Table S1.
Considering the RM190 alleles associated to the four haplotypes identified for exon 6 and the SNP at −1,514, it was observed that the combination of CT20 with A (exon 6) and G (SNP −1,514) was always associated to an AAC higher than 24.5% (Table 1), thus providing a previously un-indentified tool for selecting rice accessions with high AAC. Allelic variation observed for the SNPs at positions +1,801, +2,282 and +2,806 can finally provide useful diagnostic tools for selecting accessions with AAC higher than 24% when specific donors like Merlè, CNA 4081, Orione, Zhen Shang 47 and Alinano C are used (Table 1); for these accessions, in fact, a unique CGT allelic pattern was observed for the three SNPs. The SNP at position −1,514 slightly increased the AAC explained variation to 80.1% when considering the SNPs only, and to 80.3% when the RM190 was included (Table 3). However, when only the models having all the variables significant at P ≤ 0.05 were considered, the model with the highest ability to explain AAC variation (79.5%) was the one with SNPs at intron1 and exon 6 (Table 3), a result which is fully in agreement with previous work (Caffagni et al. 2013).
Relationships between grain shape parameters and Waxy haplotypes
Correlation analyses between grain shape parameters for the 126 non-glutinous rice accessions (Table 1) are indicated in Figure 4 A, B and showed a close agreement with those reported by Tran et al. (2012). The most obvious correlations were between the L/W ratio and either grain length (positive) or grain width (negative). A negative correlation exists between the length and the width of the grain while regarding AAC, grain length and the L/W ratio were positively correlated with it, whereas grain width was negatively correlated. An analysis of the histogram distribution plots of the variables indicated that the distributions of AAC and grain width clearly showed two modes (Figure 4 A).

Correlations between grain shape parameters and AAC. A) Scatterplot matrix of grain (dehulled caryopses, i.e., brown rice) characters. Histogram distribution plots for the single variables are shown on the diagonal cells. Below them the bivariate distributions are shown for every pair of traits. Confidence ellipses (ELL) mark confidence limit for each distribution (P = 0.95, for a normal bivariate); B) Pearson’s correlation coefficients for grain (dehulled caryopses, i.e., brown rice) characters. All the correlations are significant with P ≤ 0.001 (probabilities were adjusted by Bonferroni’s correction for multiple tests; n = 126).
In rice, traits related to grain size, shape and cooking properties have a large impact on market appreciation and play a pivotal role in the adoption of new varieties (Webb 1991; Juliano 2003). It is therefore interesting to note that, as shown in Table 6, we found some surprising associations between the haplotypes at the waxy locus and the shape of the rice caryopsis. In particular, the SNP at the first intron (which identifies Wxa and Wxb genotypes) was associated with differences in the width of the grain, the length to width ratio (L/W), and the length of the kernel. The SNP at the sixth intron showed no association with these traits by itself, but it slightly increased the overall explained variance once the SNP at the first intron was considered. This suggests that the latter SNP was the actual responsible of the association, whereas the SNP at the intron 6 has only an ancillary effect. Even the CTn showed a significant association, but its explanatory capability of the variance of grain biometric parameters was lower than that of the SNP at the first intron, thus that, given it also has no direct effect on the functionality of GBSSI, its association is most probably indirect and most likely ascribable to the phylogenetic association of some CTn haplotypes with the two versions of the SNP at the first intron.
The positive correlation between AAC and L/W ratio, and the negative one with the width of the grain were further analysed (Figure 5). In fact, by plotting L/W ratio versus AAC for the overall genotype set used in this work, the positive correlation (r = 0.517, P < 0.001) is immediately apparent (Figure 5A). When the genotype set is grouped according to the haplotype at the SNP at intron 1 (G/T), that is, the Wxa/Wxb allelic version is superimposed onto the correlation plot, it becomes evident that: (a)- this SNP offers a sharp distinction between genotypes with AAC ≤ 21% (Wxb) and AAC ≥ 21% (Wxa); (b)- in our set of genotypes, there seems to be a break for AAC around 21.5%, which, therefore, appears to be a more precise threshold value for discriminating between Wxa/Wxb allelic versions, even though a few genotypes spill over, and small differences in AAC can actually occur because of the method of assay (Fitzgerald et al. 1995; Bligh et al. 1998; Cai et al. 1958), with modifications by Inatsu (1988), the AAC of milled grain was measured with a FOSS FIAstar 5000 auto-analyzer which is based on a flow injection of a solution of NaOH 0.09% to the sample, the addition of an iodine solution and the spectrometric determination of the absorbance of the formed color at 720 nm. The calibration was performed measuring the absorbance of standard rice samples carrying 15.40%, 23.10% and 27.7% of AAC, respectively, using a white reference. These reference samples were supplied and certified by FOSS as having their amylose content determined against amylose/amylopectin standards. The SoFIA software (FOSS) was used to build up the calibration curve and to obtain the percentage of amylose in our samples. Each reference analysis was repeated twice for each sample. Biometric parameters of rice seeds (decorticated grain length and decorticated grain width) were evaluated trough optical scanner-produced high resolution images analyzed with the WinSEEDLE 2011a software (Regent Instruments Inc.).
Molecular markers analyses
To obtain genomic DNA from the rice accessions, seeds were germinated in petri dishes at 30°C and one-week old seedlings were transplanted and grown in a greenhouse until three leaf stages; leaves were therefore collected, frozen in liquid nitrogen and store at −80°C. Genomic DNA was extracted on plates using the Wizard® Magnetic 96 DNA Plant System (Promega) according to manufacturer’s instructions. The CTAB DNA extraction method (Doyle and Doyle 1987) was instead applied for the 21 genotypes selected for GBSSI gene re-sequencing.
The RM190 CT repeat was assayed using the M13-tailed forward primer RM-190 F (CACGACGTTGTAAAACGACCTTTGTCTATCTCAAGACAC) and the reverse primer RM-190R (TTGCAGATGTTCTTCCTGATG) (Ayres et al. 1997; Chen et al. 2008). PCR reactions were performed in 10 μl containing 15 ng of genomic DNA, 0.1 μM of RM-190 F, 1 μM of RM-190R and FAM-labelled M13 (CACGACGTTGTAAAACGAC), 0.2 mM dNTPs and 1 U GoTaq DNA Polymerase (Promega). DNA was amplified using a touchdown program as follows: denaturation at 94°C per 3 min, 20 cycles at 94°C per 45 sec, from 61°C to 51.5°C per 45 sec, reducing the annealing temperature of 0.5°C for each cycle, and 72°C per 45 sec, 24 cycles at 94°C per 45 sec, 51°C per 45 sec and 72°C per 45 sec and a final extension of 72°C per 10 min. Labelled PCR products were run in a 3130 Genetic Analyzer (Applied Biosystems). ROX size standard (Applied Biosystems) was used.
PCRs for dCAPS analyses of the G/T polymorphism in intron 1 were carried out in 20 μl using GoTaq DNA Polymerase (Promega) supplemented with 5% DMSO and with 20 ng of genomic DNA, 1 μM of GBSS-W2F, 1 μM of GBSS-W2R (Ayres et al. 1997; Chen et al. 2008). Amplification conditions were: 94°C per 4 min followed by 40 cycles of 94°C per 40 sec, 60°C per 50 sec and 72°C per 1 min per kb and a final extension of 72°C per 10 min. After amplification 5 μl of PCR products were digested with 1 U of AccI restriction enzyme (New England BioLabs) in a total volume of 10 μl at 37°C over night. The samples were run on 2% agarose gel. Each digested sample was compared alongside with its not-digested cognate control.
The presence of Wxop, Wxin, Wxmq, Wxhp and wx alleles and the variability for the SNP in exon 10 identified by Larking and Park (2003) were assessed in our collection through the sequencing of exons 4, 6, 5, 2 and 10 using the same procedure for the allele mining experiment described below.
To detect the SNPs at position −1,514, +1,801, +2,282 and +2,806 by dCAPS analysis, mismatched forward primers were designed by dCAPS Finder 2.0 software (http://helix.wustl.edu/dcaps/dcaps.html). Primers and restriction enzymes for dCAPS assay are listed in Additional file 1: Table S1. For PCR amplification, the same protocol used for detecting the G/T polymorphism in the leader intron was used.
Allele mining
For the allele mining of the GBSSI alleles in the 21 selected genotypes, six overlap** regions, ranging from 800 bp to 2,500 bp, were PCR amplified from genomic DNA with the same protocol described above for the G/T polymorphism. Primers designed on the Nipponbare genomic sequence (Genebank AC NC_008399; Additional file 2: Table S2) were utilized for genomic DNA amplifications using the combinations indicated in Additional file 2: Table S2. The amplified regions covered the entire gene plus 1 kbp of the upstream putative regulatory region. After gel purification by the Wizard® SV Gel and PCR Clean-Up System (Promega), PCR products were directly sequenced. Sequencing reactions were accomplished by the use of ABI BigDye Terminator version 3.1 (Applied Biosystem) in forward and reverse directions with 5 μl of each PCR amplification product and the same primers used for PCR amplifications or internal primers. All primers were designed using the Primer3 0.4.0 software (http://frodo.wi.mit.edu/) and blasted against the rice genomic sequence on the Gramene website (http://www.gramene.org) to ensure the specificity for the GBSSI gene.
Computational analyses
The RM190 microsatellite was analyzed using the GeneMapper software (Applied Biosystem). Sequence assembly was assessed with the ContigExpress tool of Vector NTI Software (Invitrogen) using the Nipponbare genomic sequence as reference. Sequence comparison was carried out by MultAlin software (http://multalin.toulouse.inra.fr/multalin/). All data were analysed with the Systat 12 software (SPSS Inc., Chicago, IL, USA). The relationships between numerical variables (AAC and grain biometrical characters) were evaluated by Pearson correlation coefficients and regression analysis. The associations between numerical variables (AAC) and categorical variables (marker haplotypes) were analysed according to the General Linear Model (GLM) procedure.
References
Asaoka M, Okuno K, Fuwa H: Effect of environmental-temperature at the milky stage and amylose content and fine-structure of amylopectin of waxy and non-waxy endosperm starches of rice (Oryza sativa L.). Agric Biol Chem 1985, 49: 373–379. 10.1271/bbb1961.49.373
Ayres NM, McClung AM, Larkin PD, Bligh HFJ, Jones CA, Park WD: Microsatellites and a single-nucleotide polymorphism differentiate apparent amylase classes in an extended pedigree of US rice germplasm. Theor Appl Genet 1997, 94: 773–781. 10.1007/s001220050477
Bao JS, Corke H, Sun M: Nucleotide diversity in starch synthase IIa and validation of single nucleotide polymorphisms in relation to starch gelatinization temperature and other physicochemical properties in rice ( Oryza sativa L.). Theor Appl Genet 2006, 113: 1171–1183. 10.1007/s00122-006-0355-6
Bennett GL, Swiger LA: Genetic variance and correlation after selection for two traits by index, independent culling levels and extreme selection. Genetics 1980, 94: 763–775.
Bergman CJ, Delgado JT, McClung AM, Fjellstrom RG: An improved method for using a microsatellite in the rice waxy gene to determine amylose class. Cereal Chem 2001, 78: 257–260. 10.1094/CCHEM.2001.78.3.257
Bligh HFG, Till RJ, Jones CA: A microsatellite sequence closely linked to the Waxy gene of Oryza sativa. Euphytica 1995, 86: 83–85. 10.1007/BF00022012
Bligh HFJ, Larkin PD, Roach PS, Jonea PA, Fu H, Park WD: Use of alternate splicing sites in granule-bound starch synthase mRNA from low-amylose rice varieties. Plant Mol Biol 1998, 38: 407–415. 10.1023/A:1006021807799
Caffagni A, Albertazzi G, Gavina G, Ravaglia S, Gianinetti A, Pecchioni N, Milc J: Characterization of an Italian rice germplasm collection with genetic markers useful for breeding to improve eating and cooking quality. Euphytica 2013, 194(3):383–399. 10.1007/s10681-013-0954-7
Cai XL, Wang ZY, **ng YY, Zhang JL, Hong MM: Aberrant splicing of intron 1 leads to the heterogeneous 5′ UTR and decreased expression of waxy gene in rice cultivars of intermediate amylose content. Plant J 1998, 14: 459–465. 10.1046/j.1365-313X.1998.00126.x
Chen MJ, Bergman C, Pinson S, Fjellstrom R: Waxy gene haplotypes: Associations with apparent amylase content and the effect by the environment in an international rice germplasm collection. J Cereal Sci 2008, 47: 536–545. 10.1016/j.jcs.2007.06.013
Chiaramonte E, Rhazi L, Aussenac T, White RD: Amylose and amylopectin in starch by asymmetric flow field-flow fractionation with multi-angle light scattering and refractive index detection (AF4–MALS–RI). J Cereal Sci 2012, 56: 457–463. 10.1016/j.jcs.2012.04.006
Dobo M, Ayres N, Walker G, Park WD: Polymorphism in the GBSS gene affects amylose content in US and European rice germplasm. J Cereal Sci 2010, 52: 450–456. 10.1016/j.jcs.2010.07.010
Doyle J, Doyle JL: Genomic plant DNA preparation from fresh tissue – the CTAB method. Phytochem Bull 1987, 19: 11.
Ferrero A, Nguyen NV: The sustainable development of rice-based production systems in Europe. Int Rice Comm Newsl 2004, 53: 115–124.
Fitzgerald MA, Bergman CJ, Resurreccion AP, Möller J, Jimenez R, Reinke RF, Martin M, Blanco P, Molina F, Chen MH, Kuri V, Romero MV, Habibi F, Umemoto T, Jongdee S, Graterol E, Reddy KR, Zaczuk Bassinello P, Sivakami R, Rani NS, Das S, Wang YJ, Indrasari SD, Ramli A, Ahmad R, Dipti SS, **e L, Lang NT, Singh P, Toro DC, Tavasoli F, Mestres C: Addressing the dilemmas of measuring amylose in rice. Cereal Chem 2009, 86(5):492–498. 10.1094/CCHEM-86-5-0492
Gomez KA: Effect of environment on protein and amylose content of rice. In Chemical aspects of rice grain quality. Los Baños, Laguna, Philippines: International Rice Research Institute, IRRI; 1979:59–68.
Hirano HY, Sano Y: Molecular characterization of the waxy locus of rice ( Oryza sativa L.). Plant Cell Physiol 1991, 32: 989–997.
Huang R, Jiang L, Zheng J, Wang T, Wang H, Huang Y, Hong Z: Genetic bases of rice grain shape: so many genes, so little known. Trends Plant Sci 2013, 18: 218–226. 10.1016/j.tplants.2012.11.001
Inatsu O: Studies on improving the eating quality of Hokkaido Rice. Report of Hokkaido Prefectural Agric. Exp Stn 1988, 66: 3–7.
Isshiki M, Morino K, Nakajima M, Okagaki RJ, Wessler SR, Izawa T, Shimamoto K: A naturally occurring functional allele of the rice waxy locus has GT to TT mutation at the 5′ splice site of the first intron. Plant J 1998, 15(1):133–138. 10.1046/j.1365-313X.1998.00189.x
Juliano BO: A simplified assay for milled-rice amylose. J Cereal Sci 1971, 16: 334–340.
Juliano BO: The chemical basis of rice grain quality. In Chemical Aspects of Rice Grain Quality. Los Baños, Laguna, Philippines: International Rice Research Institute, IRRI; 1979:69–90.
Juliano BO: Structure and function of the rice grain and its fractions. Cereal Foods World 1992, 37: 772–774.
Juliano BO: Rice chemistry and quality. Manila, Philippines: Philippine Rice Research Institute; 2003.
Kawagoe Y, Kubo A, Satoh H, Takaiwa F, Nakamura Y: Roles of isoamylase and ADP-glucose pyrophosphorylase in starch granule synthesis in rice endosperm. Plant J 2005, 42: 164–174. 10.1111/j.1365-313X.2005.02367.x
Kharabian A: An efficient computational method for screening functional SNPs in plants. J Theor Biol 2010, 265: 55–62. 10.1016/j.jtbi.2010.04.017
Kumar I, Khush GS: Genetic-analysis of different amylose levels in rice. Crop Sci 1987, 27: 1167–1172. 10.2135/cropsci1987.0011183X002700060016x
Larkin PD, Park WD: Transcript accumulation and utilization of alternate and non consensus splice sites in rice granule bound starch synthase is temperature-sensitive and controlled by a single nucleotide polymorphism. Plant Mol Biol 1999, 40: 719–727. 10.1023/A:1006298608408
Larkin PD, Park WD: Association of waxy gene single nucleotide polymorphisms with starch characteristics in rice ( Oryza sativa L.). Mol Breed 2003, 12: 335–339.
Li Y, Shoemaker CF, Ma J, Moon KJ, Zhong F: Structure-viscosity relationships for starches from different rice varieties during heating. Food Chem 2008, 106: 1105–1112. 10.1016/j.foodchem.2007.07.039
Liu L, Ma X, Liu S, Zhu C, Jiang L, Wang Y, Shen Y, Ren Y, Dong H, Chen L, Liu X, Zhao Z, Zhai H, Wan J: Identification and characterization of a novel Waxy allele from Yunnan rice landrace. Plant Mol Biol 2009, 71: 609–626. 10.1007/s11103-009-9544-4
McCouch SR, Kovach MJ, Sweeney M, Jiang H, Semon M: The dynamics of rice domestication: A balance between gene flow and genetic isolation. In Biodiversity in agriculture: domestication, evolution, and sustainability. Edited by: Paul G, Famula TR, Bettinger RL, Brush SB, Damania AB. New York, USA: Cambridge University Press; 2012:311–329.
McKenzie KS, Rutger JN: Genetic analysis of amylose content, alkali spreading score and grain dimension in rice. Crop Sci 1983, 23: 306–313. 10.2135/cropsci1983.0011183X002300020031x
Mikami I, Uwatoko N, Ikeda Y, Yamaguchi J, Hirano HY, Suzuki Y, Sano Y: Allelic diversification at the wx locus in landraces of Asian rice. Theor Appl Genet 2008, 116: 979–989. 10.1007/s00122-008-0729-z
Park IM, Ibáñez AM, Zhong F, Shoemaker CF: Gelatinization and pasting properties of waxy and non-waxy rice starches. Starch-Starke 2007, 59: 388–396. 10.1002/star.200600570
Sang T, Ge S: Understanding rice domestication and implications for cultivar improvement. Curr Opin Plant Biol 2013., 16: in press http://www.sciencedirect.com/science/article/pii/S1369526613000381 in press
Sato H, Suzuki Y, Sakai M, Imbe T: Molecular characterization of Wx-mq , anovel mutant gene for low-amylose content in endosperm of rice ( Oryza sativa L.). Breed Sci 64:2002, 52: 131–135. 10.1270/jsbbs.52.131
Satoh H, Shibahara K, Tokunaga T, Nishi A, Tasaki M, Hwang SK, Okita TW, Kaneko N, Fujita N, Yoshida M, Hosaka Y, Sato A, Utsumi Y, Ohdan T, Nakamura Y: Mutation of the plastidial alpha-glucan phosphorylase gene in rice affects the synthesis and structure of starch in the endosperm. Plant Cell 2008, 20: 1833–1849. 10.1105/tpc.107.054007
Shi W, Yang Y, Chen S, Xu M: Discovery of a new fragrance allele ant the development of functional markers for the breeding of fragrant rice varieties. Mol Breed 2008, 22: 185–192. 10.1007/s11032-008-9165-7
Shu QY, Wu DX, **a YW, Gao MW, Ayres NM, Larkin PD, Park WD: Microsatellite polymorphisms on the waxy gene locus and their relationship to amylose content in indica and japonica rice, Oryza sativa L. Acta Genet Sin 1999, 26: 350–358.
Sinervo B, Svensson E: Correlational selection and the evolution of genomic architecture. Heredity 2002, 89: 329–338. 10.1038/sj.hdy.6800148
Suwannaporn P, Pitiphunpong S, Champangern S: Classification of rice amylose content by discriminant analysis of physicochemical properties. Starch 2007, 59: 171–177. 10.1002/star.200600565
Takanokai N, Jiang H, Kubo T, Sweeney M, Matsumoto T, Kanamori H, Padhukasahasram B, Bustamante C, Yoshimura A, Doi K, McCouch S: Evolutionary history of GS3 , a gene conferring grain length in rice. Genetics 2009, 182: 1323–1334. 10.1534/genetics.109.103002
Tan YF, Zhang QF: Correlation of simple sequence repeat (SSR) variants in the leader sequence of the waxy gene with amylose of the grain in rice. Acta Bot Sin 2001, 43: 146–150.
Tran DS, Tran TTH, Nguyen TLH, Ha ML, Dinh BY, Kumamaru T, Satoh H: Variation on grain quality in Vietnamese rice cultivars collected from Central Vietnam. J Fac Agr Kyushu Univ 2012, 57: 365–371.
Venu RC, Sreerekha MV, Nobuta K, Belò A, Ning Y, An G, Meyers BC, Wang GL: Deep sequencing reveals the complex and coordinated transcriptional regulation of genes related to grain quality in rice cultivars. BMC Genomics 2011, 12: 190. 10.1186/1471-2164-12-190
Wanchana S, Too**da T, Tragoonrung S, Vanavichit A: Duplicated coding sequence in the waxy allele of tropical glutinous rice ( Oryza sativa L.). Plant Sci 2003, 165: 1193–1199. 10.1016/S0168-9452(03)00326-1
Wang ZY, Zheng FQ, Shen GZ, Gao JP, Snustad DP, Li MG, Zhang JL, Hong MM: The amylose content in rice endosperm is related to the post-transcriptional regulation of the waxy gene. Plant J 1995, 7(4):613–622. 10.1046/j.1365-313X.1995.7040613.x
Ward RM, Gao Q, de Bruyn H, Gilbert RG, Fitzgerald MA: Improved methods for the structural analysis of the amylose-rich fraction from rice flour. Biomacromolecules 2006, 7: 866–876. 10.1021/bm050617e
Webb BD: Rice quality and grades . In Rice Utilization Vol. II. Edited by: Luh BS. New York, NY: AVI Book Published by Van Nostrand Reinhold; 1991:89–119.
Wesley IJ, Osborne BG, Anderssen RS, Delwiche SR, Graybosch RA: Chemometric localization approach to NIR measurement of apparent amylose content of ground wheat. Cereal Chem 2003, 80: 462–467. 10.1094/CCHEM.2003.80.4.462
Williams VR, Wu WT, Tsai HY, Bates HG: Varietal differences in amylose content of rice starch. J Agric Food Chem 1958, 6(1):47–48. 10.1021/jf60083a009
Zeng D, Yan W, Liu X, Qian Q, Li J: Du , encoding a novel Prp1 protein regulates starch biosynthesis through affecting the splicing of Wxb pre-mRNA in rice ( Oryza sativa L.). Plant Mol Biol 2007, 65: 501–509. 10.1007/s11103-007-9186-3
Acknowledgements
This study was funded by grants from AGER Foundation, (RISINNOVA project grant n. 2010–2369) and MiPAAF (POLORISO project).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CB carried out the molecular genetic studies, the sequence alignment and drafted the manuscript; DC, RP, PB, SU, GO, FD participated to molecular genetic studies, the sequence alignment and determination of Apparent Amylose Content; AG participated in data analysis and helped to draft the manuscript; EL, LC participated in the design of the study and helped to draft the manuscript; GV designed the study and helped to draft the manuscript. All authors read and approved the final manuscript.
Chiara Biselli, Daniela Cavalluzzo contributed equally to this work.
Electronic supplementary material
12284_2013_75_MOESM1_ESM.docx
Additional file 1: Table S1: Markers obtained from the newly discovered SNPs. For each marker, primers, SNP position, restriction enzyme and origin of the restricted amplicon are indicated. The mutated base is in bold and underlined. (DOCX 12 KB)
12284_2013_75_MOESM2_ESM.doc
Additional file 2: Table S2: Primer combinations used to amplify the Waxy alleles and internal primers used for amplicons sequencing. (DOC 92 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Biselli, C., Cavalluzzo, D., Perrini, R. et al. Improvement of marker-based predictability of Apparent Amylose Content in japonica rice through GBSSI allele mining. Rice 7, 1 (2014). https://doi.org/10.1186/1939-8433-7-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1939-8433-7-1