
Abstract
Background
Treatment failure for esophageal carcinoma is frequently due to lymph node metastasis and invasion to neighboring organs. The aim of the present study was to investigate invasion- and metastasis-related genes in esophageal carcinoma cells in vitro and in vivo.
Methods
A metastasis model using a Matrigel invasion clonal selection approach was employed to establish a highly invasive subline EC9706-P4 from the esophageal carcinoma cell (ESCC) line EC9706. The differentially expressed genes of the subline and the parental cells determined by gene microarrays were further analyzed by RT-PCR and Western blotting.
Results
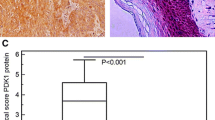
We identified sphingosine kinase 1 (SPHK1) as an invasion and metastasis-related gene of esophageal cancer. SPHK1 was overexpressed in the EC9706-P4 subline with high invasive capacity. Among six ESCC lines tested, KYSE2 and KYSE30 cells showed the highest SPHK1 mRNA and protein expressions as well as the most invasive phenotype. By Western blotting, in 7/12 cases (58%), SPHK1 expression was higher in esophageal carcinomas than in the companion normal tissue. In 23/30 cases (76%), SPHK1 protein expression was upregulated in the tumors compared to matched normal tissue by immunohistochemistry (IHC). Esophageal carcinoma tissue microarray analysis indicated that SPHK1 expression correlated with the depth of tumor invasion (P < 0.0001) and lymph node metastasis (P = 0.016). By Kaplan-Meier analysis, strong SPHK1 expression was significantly associated with clinical failure (P < 0.01), suggesting the involvement of SPHK1 in aggressiveness of human esophageal carcinoma. SPHK1 overexpression significantly increased the invasiveness of EC9706 cells in vitro and also increased EC9706 cell growth and spontaneous metastasis in vivo, promoting significant increases in tumor growth, tumor burden and spontaneous lung metastasis in nude mice. SPHK1 expression significantly correlated with the expression of many EGFR pathway genes associated with invasion of cancer cells. SPHK1 protein expression also significantly correlated with the phosphorylation of EGFR.
Conclusion
In summary, our data implicate SPHK1 in the metastasis of esophageal cancer. Our study also identified downstream mediators of SPHK1 in esophageal cancer cells that may mediate enhanced malignant behavior, and several of these mediators may be useful as therapeutic targets.
Similar content being viewed by others


Introduction
Human esophageal carcinoma, one of the most common causes of cancer death worldwide, occurs at a very high frequency in China [1, 2]. Esophageal carcinomas often have poor prognosis due to early lymph node metastasis and invasion of neighboring organs such as the aorta, trachea, bronchus, pericardium and lung [2]. Therefore, disrupting the aggressive metastatic phenotype is essential for develo** an effective treatment for esophageal cancer. Although several molecules have been reported to contribute to the ability of esophageal carcinoma cells to metastasize and invade normal tissue, such as N-cadherin [3], TSLC1 [4] and MTA1 [36]. Implicit in these stages, invasion is the critical ability for tumor cells to metastasize [36]. During invasion, malignant cells reside on or within two major types of extracellular matrices, the basement membrane and the stromal matrix [37]. The basement membrane is one of the most important barriers against cancer cell invasion [37]. Therefore, for this study, we used Matrigel, a solubilized basement membrane preparation from the Engelbreth-Holm-Swarm (EHS) mouse sarcoma, as a model basement matrix to mimic esophageal carcinoma invasion in vivo. Although EC9706, an esophageal squamous carcinoma cell line, can invade and form spontaneous lung metastasis nodules in nu/nu mice, its metastatic potential is relatively low [38]. The metastatic ability of EC9706 may arise from a few subclones with high metastatic potential among the parental cells. By screening with our in vitro model, the subline EC9706-P4 with high invasion potential was established. This subline also exhibited high spontaneous metastatic potential in vivo. Microarray analysis was used to determine which genes may be involved in invasion and metastasis. However, the microarray analysis of esophageal cancer tissues demonstrated that SPHK1 was significantly overexpressed in these tumor tissues, and that this expression significantly correlated with tumor invasion, lymph node metastasis and clinical stage, indicating that SPHK1 is involved in esophageal carcinoma invasion and metastasis. SPHK1 is up-regulated in many types of cancers and has been suggested as a potentially new therapeutic target. However, it is not yet known what signals the cancer cells use to apparently constitutively up-regulate expression of this enzyme, nor is it clear why it has such a profound role in tumorigenesis. A recent study examined the role of SPHK1 in intestinal tumorigenesis in the Min mouse in which intestinal adenomas develop spontaneously [39]. Deletion of the SPHK1 gene in these mice resulted in reduction of adenoma size. Concomitantly, epithelial cell proliferation in the polyps was attenuated, suggesting that SPHK1 regulates adenoma progression [39].
Exogenous expression of SPHK1 in vitro and in vivo further showed that it is a key factor in esophageal carcinoma cell invasion. In the transwell invasion assay, upregulation of SPHK1 expression significantly increased the invasion of EC9706 cells. Furthermore, upregulation of SPHK1 expression significantly increased the proliferation of EC9706 cells in vitro as well as increased EC9706 cell growth and spontaneous metastasis in nude mice. These studies support the view that SPHK1 expression is vital for the maintenance of invasive and metastatic potential of esophageal carcinoma cells. Interestingly, neutralizing S1P, the product of SPHK1 enzymatic activity, with a specific monoclonal antibody was remarkably effective in slowing progression of cancers, such as lung [40], colon [41], breast [42, 43], melanoma [44] and ovarian cancers [21, 45] in murine xenograft and allograft models [46]. A critical question raised by these observations is how neutralization of this simple lysophospholipid can have such dramatic effects on tumor progression.
To glean mechanistic insight into the role of SPHK1 in invasion and metastasis, we surveyed potential links between SPHK1 and key molecules related to EGFR. Western blot analysis indicated that the expression of SPHK1 was significantly correlated with the phosphorylation of EGFR. In clustering the upregulated genes in the SPHK1 overexpression clones compared with control cells, SPHK1 expression was significantly correlated with that of IL6, ITGA2, IL8, EREG, MMP1, ITGA5, MMP3 and AREG. Western blotting indicated that the ligands of EGFR, EGF, EREG and AREG induced the expression of SPHK1 protein. These findings provide evidence for cross-talk between SPHK1 and the EGFR pathway and reveal a key role for SPHK1 in integrating events downstream of EGF receptors. An intriguing possibility is that many growth and angiogenic factors such as EGF and AREG involved in tumorigenesis may act through SPHK1 activation [47]. Because both EGF and AREG have been implicated in progression of esophageal cancer, it was of interest to examine the involvement of SPHK1. There are numerous reports of rapid and transient activation of SPHK1 by growth and angiogenic factors [48, 49]) that stimulate its phosphorylation at Ser225 [50] and subsequent translocation to the plasma membrane [51] where its substrate sphingosine resides, resulting in local formation of S1P [52]. Some cross-talk models between EGFR and SPHK1/S1P have been proposed previously, Estrada-Bernal, A et al[53] have reported that treatment of glioma cell lines with EGF led to increased expression and activity of SphK1. Expression of EGFRvIII in glioma cells also activated and induced SphK1. In addition, siRNA to SphK1 partially inhibited EGFRvIII-induced growth and survival of glioma cells as well as ERK MAP kinase activation. SphK1 activity is necessary for survival of GBM-derived neurosphere cells, and EGFRvIII partially utilizes SphK1 to further enhance cell proliferation. Shida, D. et al[54]reported that LPA markedly enhanced SphK1 mRNA and protein in gastric cancer MKN1 cells, DLD1 colon cancer cells and MDA-MB-231 breast cancer cells. LPA transactivated the epidermal growth factor receptor (EGFR) in these cells, and the EGFR inhibitor AG1478 attenuated the increased SphK1 and S1P(3) expression induced by LPA. Their research finally showed that SphK1 is a convergence point of multiple cell surface receptors for three different ligands, LPA, EGF, and S1P, which have all been implicated in regulation of motility and invasiveness of cancer cells. In breast cancer, Sukocheva, O et al[55]demonstrated that E2-induced EGFR transactivation in human breast cancer cells is driven via a novel signaling system controlled by the lipid kinase sphingosine kinase-1 (SphK1). E2 stimulates SphK1 activation and the release of sphingosine 1-phosphate (S1P), by which E2 is capable of activating the S1P receptor Edg-3, resulting in the EGFR transactivation in a matrix metalloprotease-dependent manner. These findings reveal a key role for SphK1 in the coupling of the signals between three membrane-spanning events induced by E2, S1P, and EGF. However, it is still difficult to understand how such short-lived activation can be responsible for the profound involvement of SPHK1 in tumorigenicity or how this relates to its up-regulation in cancer. Our results imply that SPHK1 may be the central controller of amplification loops of EGF, AREG and EREG-EGFR interactions that can contribute to cancer progression.
Conclusions
We have established an esophageal carcinoma invasion model and generated a highly invasive tumor cell subline in which SPHK1 was overexpressed. Further investigation revealed SPHK1 was significantly correlated with esophageal cancer invasion and metastasis and may be a valuable prognostic marker. Our studies have demonstrated that SPHK1 is involved in upregulation of EREG and AREG through enhancing EGFR phosphorylation to promote invasion. Thus, modulating SPHK1 expression or activity is an attractive additional therapeutic strategy for treatment of esophageal cancer and perhaps other cancers as well. Implementation of pre-clinical and clinical evaluation of SPHK1 as a novel molecular target for cancer therapy is warranted.
Authors' information
Pan Jian, Ph.D. Immunology. Graduated from State Key Laboratory of Molecular Oncology, Cancer Institute (Hospital), Peking Union Medical College, Chinese Academy of Medical Sciences, Bei**g, PR China. Now is an associate professor of Department of Hematology and Oncology, Children's Hospital of Soochow University, Suzhou, China, and a gust professor of Translational Research Center, Second Hospital, The Second Clinical School, Nan**g Medical University, Nan**g, China.
References
Maddams J, Parkin DM, S CD: The cancer burden in the UK in 2007 due to radiotherapy. Int J Cancer.
Enzinger PC, Mayer RJ: Esophageal cancer. N Engl J Med. 2003, 349: 2241-2252. 10.1056/NEJMra035010.
Yoshinaga K, Inoue H, Utsunomiya T, Sonoda H, Masuda T, Mimori K, Tanaka Y, Mori M: N-cadherin is regulated by activin A and associated with tumor aggressiveness in esophageal carcinoma. Clin Cancer Res. 2004, 10: 5702-5707. 10.1158/1078-0432.CCR-03-0262.
Ito T, Shimada Y, Hashimoto Y, Kaganoi J, Kan T, Watanabe G, Murakami Y, Imamura M: Involvement of TSLC1 in progression of esophageal squamous cell carcinoma. Cancer Res. 2003, 63: 6320-6326.
Qian H, Lu N, Xue L, Liang X, Zhang X, Fu M, **e Y, Zhan Q, Liu Z, Lin C: Reduced MTA1 Expression by RNAi Inhibits in Vitro Invasion and Migration of Esophageal Squamous Cell Carcinoma Cell Line. Clin Exp Metastasis. 2006
Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, Shao R, Anderson RM, Rich JN, Wang XF: Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. 2004, 5: 329-339. 10.1016/S1535-6108(04)00081-9.
Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massague J: Genes that mediate breast cancer metastasis to lung. Nature. 2005, 436: 518-524. 10.1038/nature03799.
Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J: A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003, 3: 537-549. 10.1016/S1535-6108(03)00132-6.
Gupta GP, Nguyen DX, Chiang AC, Bos PD, Kim JY, Nadal C, Gomis RR, Manova-Todorova K, Massague J: Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature. 2007, 446: 765-770. 10.1038/nature05760.
Chen XL, Grey JY, Thomas S, Qiu FH, Medford RM, Wasserman MA, Kunsch C: Sphingosine kinase-1 mediates TNF-alpha-induced MCP-1 gene expression in endothelial cells: upregulation by oscillatory flow. Am J Physiol Heart Circ Physiol. 2004, 287: H1452-1458. 10.1152/ajpheart.01101.2003.
Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T: Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell Signal. 2005, 17: 1203-1217. 10.1016/j.cellsig.2004.12.005.
Bu S, Yamanaka M, Pei H, Bielawska A, Bielawski J, Hannun YA, Obeid L, Trojanowska M: Dihydrosphingosine 1-phosphate stimulates MMP1 gene expression via activation of ERK1/2-Ets1 pathway in human fibroblasts. Faseb J. 2006, 20: 184-186.
Bonhoure E, Lauret A, Barnes DJ, Martin C, Malavaud B, Kohama T, Melo JV, Cuvillier O: Sphingosine kinase-1 is a downstream regulator of imatinib-induced apoptosis in chronic myeloid leukemia cells. Leukemia. 2008, 22: 971-979. 10.1038/leu.2008.95.
Martin C, Lafosse JM, Malavaud B, Cuvillier O: Sphingosine kinase-1 mediates androgen-induced osteoblast cell growth. Biochem Biophys Res Commun. 391: 669-673.
Kapitonov D, Allegood JC, Mitchell C, Hait NC, Almenara JA, Adams JK, Zipkin RE, Dent P, Kordula T, Milstien S, Spiegel S: Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009, 69: 6915-6923. 10.1158/0008-5472.CAN-09-0664.
Young N, Pearl DK, Van Brocklyn JR: Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res. 2009, 7: 23-32. 10.1158/1541-7786.MCR-08-0061.
Ran Y, Pan J, Hu H, Zhou Z, Sun L, Peng L, Yu L, Sun L, Liu J, Yang Z: A novel role for tissue factor pathway inhibitor-2 in the therapy of human esophageal carcinoma. Hum Gene Ther. 2009, 20: 41-49. 10.1089/hum.2008.129.
Pan J, Hu H, Zhou Z, Sun L, Peng L, Yu L, Sun L, Liu J, Yang Z, Ran Y: Tumor-suppressive mir-663 gene induces mitotic catastrophe growth arrest in human gastric cancer cells. Oncol Rep. 24: 105-112.
Jian P, Yanfang T, Zhuan Z, Jian W, Xueming Z, Jian N: MMP28 (epilysin) as a novel promoter of invasion and metastasis in gastric cancer. BMC Cancer. 11: 200-
Jian P, Li ZW, Fang TY, Jian W, Zhuan Z, Mei LX, Yan WS, Jian N: Retinoic acid induces HL-60 cell differentiation via the upregulation of miR-663. J Hematol Oncol. 4: 20-
Guillermet-Guibert J, Davenne L, Pchejetski D, Saint-Laurent N, Brizuela L, Guilbeau-Frugier C, Delisle MB, Cuvillier O, Susini C, Bousquet C: Targeting the sphingolipid metabolism to defeat pancreatic cancer cell resistance to the chemotherapeutic gemcitabine drug. Mol Cancer Ther. 2009, 8: 809-820. 10.1158/1535-7163.MCT-08-1096.
Barker HE, Chang J, Cox TR, Lang G, Bird D, Nicolau M, Evans HR, Gartland A, Erler JT: LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Res. 71: 1561-1572.
Peng L, Ran YL, Hu H, Yu L, Liu Q, Zhou Z, Sun YM, Sun LC, Pan J, Sun LX: Secreted LOXL2 is a novel therapeutic target that promotes gastric cancer metastasis via the Src/FAK pathway. Carcinogenesis. 2009, 30: 1660-1669. 10.1093/carcin/bgp178.
Schietke R, Warnecke C, Wacker I, Schodel J, Mole DR, Campean V, Amann K, Goppelt-Struebe M, Behrens J, Eckardt KU, Wiesener MS: The lysyl oxidases LOX and LOXL2 are necessary and sufficient to repress E-cadherin in hypoxia: insights into cellular transformation processes mediated by HIF-1. J Biol Chem. 285: 6658-6669.
Mochizuki S, Okada Y: ADAMs in cancer cell proliferation and progression. Cancer Sci. 2007, 98: 621-628. 10.1111/j.1349-7006.2007.00434.x.
Roy R, Rodig S, Bielenberg D, Zurakowski D, Moses MA: ADAM12 transmembrane and secreted isoforms promote breast tumor growth: a distinct role for ADAM12-S protein in tumor metastasis. J Biol Chem. 286: 20758-20768.
Chakraborty C, Gleeson LM, McKinnon T, Lala PK: Regulation of human trophoblast migration and invasiveness. Can J Physiol Pharmacol. 2002, 80: 116-124. 10.1139/y02-016.
Irwin JC, Suen LF, Martina NA, Mark SP, Giudice LC: Role of the IGF system in trophoblast invasion and pre-eclampsia. Hum Reprod. 1999, 14 (Suppl 2): 90-96.
Demetriou MC, Kwei KA, Powell MB, Nagle RB, Bowden GT, Cress AE: Integrin A6 Cleavage in Mouse Skin Tumors. Open Cancer J. 2008, 2: 1-4.
Goplen D, Wang J, Enger PO, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R: Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res. 2006, 66: 9895-9902. 10.1158/0008-5472.CAN-05-4589.
Hori T, Yamashita Y, Ohira M, Matsumura Y, Muguruma K, Hirakawa K: A novel orthotopic implantation model of human esophageal carcinoma in nude rats: CD44H mediates cancer cell invasion in vitro and in vivo. Int J Cancer. 2001, 92: 489-496. 10.1002/ijc.1234.
Le Scolan E, Pchejetski D, Banno Y, Denis N, Mayeux P, Vainchenker W, Levade T, Moreau-Gachelin F: Overexpression of sphingosine kinase 1 is an oncogenic event in erythroleukemic progression. Blood. 2005, 106: 1808-1816. 10.1182/blood-2004-12-4832.
Li XD, Miao SY, Wang GL, Yang L, Shu YQ, Yin YM: Amphiregulin and epiregulin expression in colorectal carcinoma and the correlation with clinicopathological characteristics. Onkologie. 33: 353-358.
Orso F, Penna E, Cimino D, Astanina E, Maione F, Valdembri D, Giraudo E, Serini G, Sismondi P, De Bortoli M, Taverna D: AP-2alpha and AP-2gamma regulate tumor progression via specific genetic programs. Faseb J. 2008, 22: 2702-2714. 10.1096/fj.08-106492.
Garamszegi N, Garamszegi SP, Scully SP: Matrix metalloproteinase-1 contribution to sarcoma cell invasion. J Cell Mol Med.
Fidler IJ: The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003, 3: 453-458. 10.1038/nrc1098.
Even-Ram S, Yamada KM: Cell migration in 3D matrix. Curr Opin Cell Biol. 2005, 17: 524-532. 10.1016/j.ceb.2005.08.015.
Lan T, Shen X, Liu P, Liu W, Xu S, **e X, Jiang Q, Li W, Huang H: Berberine ameliorates renal injury in diabetic C57BL/6 mice: Involvement of suppression of SphK-S1P signaling pathway. Arch Biochem Biophys. 502: 112-120.
Kohno M, Momoi M, Oo ML, Paik JH, Lee YM, Venkataraman K, Ai Y, Ristimaki AP, Fyrst H, Sano H: Intracellular role for sphingosine kinase 1 in intestinal adenoma cell proliferation. Mol Cell Biol. 2006, 26: 7211-7223. 10.1128/MCB.02341-05.
Song L, **ong H, Li M, Liao WT, Wang L, Wu J: Sphingosine kinase-1 Enhances Resistance to Apoptosis through Activation of PI3K/Akt/NF-{kappa}B Pathway in Human Non-small Cell Lung Cancer. Clin Cancer Res.
Nemoto S, Nakamura M, Osawa Y, Kono S, Itoh Y, Okano Y, Murate T, Hara A, Ueda H, Nozawa Y, Banno Y: Sphingosine kinase isoforms regulate oxaliplatin sensitivity of human colon cancer cells through ceramide accumulation and Akt activation. J Biol Chem. 2009, 284: 10422-10432. 10.1074/jbc.M900735200.
Sukocheva O, Wang L, Verrier E, Vadas MA, **a P: Restoring endocrine response in breast cancer cells by inhibition of the sphingosine kinase-1 signaling pathway. Endocrinology. 2009, 150: 4484-4492. 10.1210/en.2009-0391.
Sarkar S, Maceyka M, Hait NC, Paugh SW, Sankala H, Milstien S, Spiegel S: Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005, 579: 5313-5317. 10.1016/j.febslet.2005.08.055.
Maceyka M, Alvarez SE, Milstien S, Spiegel S: Filamin A links sphingosine kinase 1 and sphingosine-1-phosphate receptor 1 at lamellipodia to orchestrate cell migration. Mol Cell Biol. 2008, 28: 5687-5697. 10.1128/MCB.00465-08.
Elloul S, Vaksman O, Stavnes HT, Trope CG, Davidson B, Reich R: Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin Exp Metastasis. 27: 161-172.
Visentin B, Vekich JA, Sibbald BJ, Cavalli AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS: Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell. 2006, 9: 225-238. 10.1016/j.ccr.2006.02.023.
Milstien S, Spiegel S: Targeting sphingosine-1-phosphate: a novel avenue for cancer therapeutics. Cancer Cell. 2006, 9: 148-150. 10.1016/j.ccr.2006.02.025.
Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S: Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006, 1758: 2016-2026. 10.1016/j.bbamem.2006.08.007.
Taha TA, Hannun YA, Obeid LM: Sphingosine kinase: biochemical and cellular regulation and role in disease. J Biochem Mol Biol. 2006, 39: 113-131. 10.5483/BMBRep.2006.39.2.113.
Pitson SM, Moretti PA, Zebol JR, Lynn HE, **a P, Vadas MA, Wattenberg BW: Activation of sphingosine kinase 1 by ERK1/2-mediated phosphorylation. Embo J. 2003, 22: 5491-5500. 10.1093/emboj/cdg540.
Stahelin RV, Hwang JH, Kim JH, Park ZY, Johnson KR, Obeid LM, Cho W: The mechanism of membrane targeting of human sphingosine kinase 1. J Biol Chem. 2005, 280: 43030-43038. 10.1074/jbc.M507574200.
Spiegel S, Milstien S: Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003, 4: 397-407. 10.1038/nrm1103.
Estrada-Bernal A, Lawler SE, Nowicki MO, Ray Chaudhury A, Van Brocklyn JR: The role of sphingosine kinase-1 in EGFRvIII-regulated growth and survival of glioblastoma cells. J Neurooncol. 102: 353-366.
Shida D, Fang X, Kordula T, Takabe K, Lepine S, Alvarez SE, Milstien S, Spiegel S: Cross-talk between LPA1 and epidermal growth factor receptors mediates up-regulation of sphingosine kinase 1 to promote gastric cancer cell motility and invasion. Cancer Res. 2008, 68: 6569-6577. 10.1158/0008-5472.CAN-08-0411.
Sukocheva O, Wadham C, Holmes A, Albanese N, Verrier E, Feng F, Bernal A, Derian CK, Ullrich A, Vadas MA, **a P: Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: the role of sphingosine kinase-1. J Cell Biol. 2006, 173: 301-310. 10.1083/jcb.200506033.
Acknowledgements
We thank Professor Zhihua Yang (Cancer Institute/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College, Bei**g, China) for her kind help. We thank Dr. Mingrong Wang (Cancer Institute/Cancer Hospital, Chinese Academy of Medical Sciences & Peking Union Medical College, Bei**g, China) for kindly providing the EC9706 cell line and Dr. Shimada Y (Kyoto University Graduate School of Medicine, Japan) for the KYSE esophageal cancer cells.
This work was supported by grants from the Natural Science Foundation for the Youth (No.81100371), National Key Basic Research Program (NKBRP) (973 program) (No.2010CB933902) and the National Natural Science Foundation (30570818 and 30600279).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PJ and NJ designed the study and wrote the manuscript, FX and ZWL participated in data analysis, PJ, WJ, ZYL and TYF finished the most experiment, HSY and WSY performed flow cytometry analysis. CBR and LZ collected the samples and made great contribution in making the tissue microarray. All authors read and approved the final manuscript.
Electronic supplementary material
12967_2011_822_MOESM1_ESM.XLS
Additional file 1: The gene expression profiles of EC9706 and EC9706-P4 were analyzed by microarrays. Between these cell lines, 124 genes were differentially expressed, including 71 upregulated genes and 53 downregulated genes in EC9706-P4 relative to EC9706 (cutoff, fold ≥ 2.0). (XLS 30 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pan, J., Tao, YF., Zhou, Z. et al. An novel role of sphingosine kinase-1 (SPHK1) in the invasion and metastasis of esophageal carcinoma. J Transl Med 9, 157 (2011). https://doi.org/10.1186/1479-5876-9-157
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-9-157