
Abstract
Background
Prostate cancer that has recurred after local therapy or disseminated distantly is usually treated with androgen deprivation therapy; however, most men will eventually experience disease progression within 12 to 20 months. New data emerging from randomized controlled trials (RCTs) of chemotherapy provided the impetus for a systematic review addressing the following question: which non-hormonal systemic therapies are most beneficial for the treatment of men with hormone-refractory prostate cancer (HRPC) and clinical evidence of metastases?
Methods
A systematic review was performed to identify RCTs or meta-analyses examining first-line non-hormonal systemic (cytotoxic and non-cytotoxic) therapy in patients with HRPC and metastases that reported at least one of the following endpoints: overall survival, disease control, palliative response, quality of life, and toxicity. Excluded were RCTs of second-line hormonal therapies, bisphosphonates or radiopharmaceuticals, or randomized fewer than 50 patients per trial arm. MEDLINE, EMBASE, the Cochrane Library, and the conference proceedings of the American Society of Clinical Oncology were searched for relevant trials. Citations were screened for eligibility by four reviewers and discrepancies were handled by consensus.
Results
Of the 80 RCTs identified, 27 met the eligibility criteria. Two recent, large trials reported improved overall survival with docetaxel-based chemotherapy compared to mitoxantrone-prednisone. Improved progression-free survival and rates of palliative and objective response were also observed. Compared with mitoxantrone, docetaxel treatment was associated with more frequent mild toxicities, similar rates of serious toxicities, and better quality of life. More frequent serious toxicities were observed when docetaxel was combined with estramustine. Three trials reported improved time-to-disease progression, palliative response, and/or quality of life with mitoxatrone plus corticosteroid compared with corticosteroid alone. Single trials reported improved disease control with estramustine-vinblastine, vinorelbine-hydrocortisone, and suramin-hydrocortisone compared to controls. Trials of non-cytotoxic agents have reported equivocal results.
Conclusion
Docetaxel-based chemotherapy modestly improves survival and provides palliation for men with HRPC and metastases. Other than androgen deprivation therapy, this is the only other therapy to have demonstrated improved overall survival in prostate cancer in RCTs. Further investigations to identify more effective therapies for HRPC including the use of systemic therapies earlier in the natural history of prostate cancer are warranted.
Similar content being viewed by others


Background
Prostate cancer is the most frequently diagnosed cancer in North America, and the third most common cause of cancer death in men [1]. Men with prostate cancer that has recurred after local therapy or disseminated distantly usually respond to androgen deprivation therapy (ADT). However, most patients eventually experience disease progression within a median of 12 to 20 months [2]. Androgen independence is defined as disease progression despite effective ADT, typically first identified by a rise in serum prostatic-specific antigen (PSA) levels. Hormone-refractory prostate cancer (HRPC) arises when disease progression continues despite secondary hormonal maneuvers and antiandrogen withdrawal (AAWD). The prognosis of HRPC is associated with performance status, the presence of bone pain, extent of disease on bone scan, and serum alkaline phosphatase levels [3]. Bone metastases will occur in 90% of men with HRPC and can produce significant morbidity including pain, pathologic fractures, spinal cord compression, and bone marrow failure [4]. Paraneoplastic effects are also common, including anemia, weight loss, fatigue, hypercoagulability, and increased susceptibility to infection. Thus, HRPC presents a spectrum of disease ranging from patients without metastases or symptoms with rising PSA levels despite ADT, to patients with metastases and significant debilitation due to cancer symptoms. Historically, clinical management has been primarily palliative with a focus on expectant management when possible and palliative interventions such as radiotherapy, radioisotopes, and chemotherapy when necessary [5]. New data emerging from large clinical trials of chemotherapy provided the impetus for this systematic review of the value of chemotherapy and other non-hormonal agents in HRPC.
Methods
Development of systematic review
The Genitourinary Cancer Disease Site Group of the Cancer Care Ontario Program in Evidence-Based Care (CCOPEBC) has developed an evidence-based clinical practice guideline on the use of non-hormonal systemic therapy for HRPC using the methodology outlined in the practice guideline development cycle by Browman et al [6]. The guideline was derived from a systematic review and input from practitioners in Ontario, Canada. This report describes the systematic review portion of the guideline, which addressed the following question: which non-hormonal systemic therapies are most beneficial for the treatment of men with HRPC and clinical evidence of metastases?
Inclusion criteria
Articles were selected for inclusion in the systematic review if they were randomized controlled trials (RCTs) or meta-analyses of RCTs studying a first-line non-hormonal systemic (cytotoxic and non-cytotoxic) therapy in patients with HRPC and metastases that reported on at least one of the following outcomes: overall survival, disease control (i.e., progression-free survival [PFS], time-to-progression [TTP], time-to-treatment failure, objective tumor response, and PSA response), palliative or symptomatic response, quality of life (QoL), and/or toxicity. Previous systematic reviews or evidence-based guidelines that addressed non-hormonal systemic therapy in HRPC were also eligible for inclusion.
Exclusion criteria
Articles were excluded from the systematic review if they studied second-line hormonal therapies, bisphosphonates or radiopharmaceuticals, or randomized fewer than 50 patients per trial arm. When formulating the protocol for this review, the authors were aware of dozens of small randomized trials comparing the activity of various drugs in HRPC. Sometimes these were identified explicitly as phase II trials, but often they were not. Although valuable for identifying potential anti-tumor activity, such trials are by definition underpowered to address the patient outcomes of interest to this review, and often did not report these. Although these limitations could be overcome by statistical pooling, it is also recognized that such RCTs are associated with more variability and are more likely to be reported and published if positive [7]. Theoretically, positive results from such trials require subsequent confirmation by larger pragmatic RCTs, but this does not always occur. After considering the outcomes of interest for this review, a minimum sample size of 50 randomized patients per trial arm was chosen, in order to be as inclusive as possible while still minimizing inclusion of randomized phase II trials. This sample size was based on a requirement of at least 80% power for an RCT to reliably detect the difference between an endpoint response rate of 10% versus 30% with one-tailed α = 0.05. Reliable assessment of outcomes such as disease-free and overall survival would require even larger sample sizes. RCTs with lesser discriminating ability were considered underpowered and potentially misleading with regard to those endpoints.
Literature search strategy
The MEDLINE (1966 through March 2004), EMBASE (1980 through 2004, week 10), and Cochrane Library (2003, Issue 4) databases (Central Register of Controlled Trials [CCTR] and Database of Systematic Reviews [DSR]), and the conference proceedings of the American Society of Clinical Oncology (1999 through 2004) were searched for abstracts of relevant trials. MEDLINE was searched using the following medical subject headings (MeSH): "prostatic neoplasms", "drug therapy", "antineoplastic agents", and "drug therapy, combination"; and EMBASE was searched using the following Excerpta Medica tree terms: "prostate tumor", "prostate cancer", "drug therapy", "antineoplastic agent", "drug combination", and "combination chemotherapy". In each database, those subject headings were combined with disease and treatment-specific text words (e.g., "prostate cancer", "prostate tumor", "prostate carcinoma", and "chemotherapy"). Those terms were then combined with the search terms for the following publication types and study designs: practice guidelines, systematic reviews, meta-analyses, reviews, randomized controlled trials, and controlled clinical trials. The CCTR and DSR databases were searched using a combination of the aforementioned MeSH and keywords. The reference lists from eligible articles were searched for additional trials, as were the reference lists from relevant review papers.
Methods of review
Citations identified by the literature search strategy were screened for eligibility by four of the authors (EW, DSE, SB, SH) and discrepancies were handled by consensus. Data pertaining to trial design, participants, interventions, and outcomes were extracted from each eligible trial by one reviewer (TW) and were audited by a second reviewer (EW) independently. Information indicative of trial quality, including methods of randomization, absence or degree of blinding, completeness of patient follow-up, and whether statistical analyses were performed by intent-to-treat were also extracted from each trial report. The quality parameters assessed on each trial were used to identify any trials with serious flaws in trial methodology that could lead to potentially misleading results.
Synthesis of the evidence
This review is based on data provided by published reports. When pooling of data is to be undertaken and individual patient data are not available, methods are available for generating summary statistics from published data. However, statistical pooling (meta-analysis) of data from RCTs is only possible if control arms are similar and is only valid if those data are statistically homogeneous. Reports of RCTs studying systemic therapies in HRPC date back 30 years and have studied heterogeneous patient populations, interventions, and outcomes. Numerous drug interventions have been tested, including a variety of single-agent and combination chemotherapy regimens including estramustine phosphate (EMP), and non-cytotoxic drugs such as liarazole, suramin, and atrasentan. What constitutes standard therapy in control arms has been controversial and has included placebo, corticosteroids, EMP, and cytotoxics. On the basis of those observations, large-scale quantitative pooling of RCT data was considered neither possible nor appropriate, and so an interpretive summary of the data was planned. The natural history and management of HRPC has changed in the last three decades; therefore, more contemporary studies were emphasized in the interpretive summary to provide clinicians with the evidence most relevant to current practice. Furthermore, more emphasis on the results of RCTs demonstrating internally consistent benefits in survival, palliation, and quality of life outcomes was planned.
Results
Literature search results
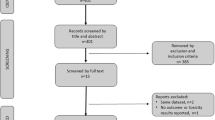
The literature search identified 80 unique RCTs that compared non-hormonal systemic treatments in HRPC. Fifty-three of those trials randomized less than 50 patients per arm and were considered ineligible [8–60] (see Additional file 1). Of the 27 eligible trials, 22 were published as full reports [61–83], and five were available only in abstract or poster presentation form but provided adequate data on at least one outcome of interest [84–88]. There were 21 two-arm and six three-arm trials, and two trials were labeled as randomized phase II reports [79, 84]. No published systematic reviews or evidence-based guidelines were identified. No trials were excluded due to their quality.
The 27 RCTs that form the basis of this review were published between 1979 and 2004. A total of 7489 eligible men were randomized, ranging from 102 to 1006 per RCT. Six trials were placebo-controlled [65, 79, 82, 85–87], and four of those were also double-blinded [65, 79, 82, 85]. Twelve trials described the methods used to randomize patients [61–65, 70–73, 79, 83, 86], and 21 reported that treatment arms were balanced for important baseline prognostic factors [61–67, 69–73, 75–79, 81–83, 87]. Twelve trials performed statistical analyses according to intent-to-treat [62–64, 70, 71, 74, 79, 81–83, 85, 86]. A minority of trials reported whether patients underwent AAWD [61–64, 69–71, 79, 81, 83] and continued to receive ADT during the study period [61–64, 69–71, 81–83]. Twenty trials studied cytotoxic [61–78, 84, 88] and seven studied non-cytotoxic drug interventions [79–83, 85–87]. Two cytotoxic trials studied agents belonging to multiple drug classes [75, 88]. For clarity, the cytotoxic trials have been organized according to the drug class tested by the trial: antimicrotubule-based regimens (nine trials), anthracendedione/anthracycline-based regimens (nine trials), and other chemotherapy (four trials).
Antimicrotubule-based chemotherapy
Docetaxel
Docetaxel induces polymerization of microtubules and phosphorylation of bcl-2 protein. Two large RCTs comparing docetaxel-based chemotherapy to mitoxantrone-prednisone have recently been published (Table 1). Tannock et al [64] randomized 1006 patients to one of three treatment arms: docetaxel (75 mg/m2 intravenously [iv] every [q] three weeks), docetaxel (30 mg/m2 iv five-times weekly for five of six weeks), or control therapy with mitoxantrone. Patients in all three arms also received prednisone (5 mg orally [po] twice daily). Petrylak et al [63] reported on 666 eligible patients randomized to docetaxel and EMP or mitoxantrone-prednisone. In addition to dexamethasone premedication, patients in the docetaxel arm also received warfarin and acetylsalicylic acid (ASA) as thrombosis prophylaxis during the course of the trial. Men in both trials had clinical evidence of metastases with or without symptoms and had undergone AAWD. Overall survival was the primary endpoint in both trials.
Tannock et al [64] reported improved survival with docetaxel-prednisone (q third week) compared with mitoxantrone-prednisone (median survival, 18.9 versus 16.5 months; hazard ratio [HR] = 0.76 [95% confidence interval (CI), 0.62–0.94], two-sided p = 0.009) (Table 2). No overall survival benefit was observed with docetaxel-prednisone given on a weekly schedule (HR = 0.91, [95% CI, 0.75–1.11], two-sided p = 0.36). Petrylak et al [63] reported longer survival time with docetaxel-EMP combination chemotherapy compared with mitoxantrone-prednisone (median survival, 17.5 versus 15.6 months; HR = 0.80 [95% CI, 0.67–0.97], two-sided p = 0.02) (Table 2). This trial also reported a median progression-free interval of 6.3 versus 3.2 months (HR = 0.73 [95% CI, 0.63–0.86], two-sided p < 0.0001) favoring docetaxel-EMP compared with mitoxantrone-prednisone.
Pain response was assessed in both trials. Significantly more patients treated with docetaxel-prednisone (q third week) achieved a pain response compared with patients receiving mitoxantrone-prednisone (35% versus 22%, p = 0.01) [64]. A trend towards improved pain response was observed with weekly docetaxel-prednisone versus mitoxantrone-prednisone (31% versus 22%, p = 0.08). QoL response defined as a sustained 16-point or greater improvement from baseline on two consecutive measurements was higher with docetaxel given every three weeks (22% versus 13%, p = 0.009) or weekly (23% versus 13%, p = 0.005) compared with mitoxantrone. Petrylak et al [63] reported no difference in patient reported pain relief between arms in their trial and did not assess QoL.
In both trials, PSA response rates were also statistically significantly higher with docetaxel compared to mitoxantrone (Table 3). Twenty-seven per cent (n = 412) [64] and 29% (n = 196) [63] of patients in the two trials had measurable disease. Objective response rates for docetaxel-prednisone (q three weeks) and mitoxantrone-prednisone were 12% versus 7%, respectively (Table 3). Petrylak et al [63] reported objective response rates of 17% and 11% favoring docetaxel-EMP compared with mitoxantrone-prednisone. The differences in objective response rates between arms were not statistically significant in either trial.
More grade 3–4 neutropenia (32% and 22% versus 1.5%) and neutropenic infection (3% and 0.9% versus 0%) were observed with docetaxel-prednisone (q third week) compared with mitoxantrone-prednisone and docetaxel-prednisone given weekly, respectively [64]. However, only two septic deaths occurred, one each in the mitoxantrone and weekly docetaxel arms. Grade 3–4 non-hematological toxicities were infrequent and similar in the docetaxel and mitoxantrone arms. Mild to moderate alopecia, fatigue, diarrhea, nail changes, stomatitis, peripheral edema, anorexia, and dyspnea were more common with docetaxel. More grade 3–4 toxicity (53% versus 33%) was associated with docetaxel-EMP compared with mitoxantrone-prednisone, primarily due to higher rates of gastrointestinal and cardiovascular events [63]. The protocol was amended to add oral coumadin (2 mg daily) and oral ASA (325 mg daily) to the docetaxel arm, but post hoc analysis suggested prophylactic anticoagulation had little effect on the rate of thromboembolic events. Docetaxel-EMP was also associated with statistically significantly higher rates of metabolic disturbances (6% versus 1%) and neurologic events (7% versus 2%) compared to mitoxantrone-prednisone. Eight (2%) versus four (1%) toxic deaths occurred in the docetaxel-EMP and mitoxantrone arms, respectively.
Estramustine
EMP is a nor-nitrogen mustard carbamate derivative of estradiol-17β-phosphate with estrogenic and antimicrotuble effects. It is unclear how much of this agent's activity in HRPC is due to its hormonal versus its cytotoxic effects. Six RCTs directly examined the efficacy of EMP in HRPC (Table 1). Three studied EMP either by comparing it to a placebo or an oral antiandrogen [65–67], and three added EMP to a cytotoxic agent and compared this combination to the cytotoxic agent alone [61, 68, 84]. One other large RCT comparing docetaxel-EMP to mitoxantrone-prednisone could be considered to indirectly address the value of EMP (see Docetaxel above) [63].
All six trials reported on overall survival, but none detected improvements with EMP (Table 2). Five trials reported TTP or PFS results; of those, one trial comparing EMP-vinblastine to vinblastine alone reported longer TTP with the combination (median, 3.7 versus 2.2 months, one-sided p < 0.0004) [61]. EMP was not associated with improved pain, performance status, or subjective response rate in two trials reporting those data [65, 68]. Hudes et al [61] reported improved pain frequency with EMP; however, less than 50% of patients with pain completed pain questionnaires. QoL data were also collected in that trial but did not allow comparative assessment. The three RCTs reporting on PSA response showed higher PSA response rates with EMP [61, 65, 84] (Table 3). Three RCTs reported objective response rates [61, 67, 68] and none showed improved tumor response with EMP (Table 3).
EMP was generally associated with clinically significant higher rates and severity of gastrointestinal toxicity (including nausea and vomiting, diarrhea, and dyspepsia), breast tenderness/gynecomastia, leg edema, thrombosis, and cardiovascular deaths [61, 65–67]. The addition of EMP to chemotherapy was also associated with a reduced incidence and severity of neutropenia [68].
Vinorelbine
Vinorelbine is a semi-synthetic vinca alkaloid with single-agent activity in HRPC. Abratt et al [62] randomized 414 men treated with hydrocortisone with or without aminoglutethimide to vinorelbine or no chemotherapy (Table 1). The primary endpoint of the trial was PFS. A longer progression-free interval was reported with vinorelbine after adjustment for predetermined prognostic factors (median, 3.7 versus 2.8 months; HR = 0.71, unadjusted two-sided p = 0.055, adjusted two-sided p = 0.005). No difference in overall survival was detected (Table 2). Thirty-four percent of patients (n = 142) had measurable disease and response rates of 5.9% (partial only) and 0% were reported favoring vinorelbine (p-value not reported) (Table 3). PSA response rates (Table 3) and clinical benefit response (defined as improved pain, analgesic score, or performance status for greater than nine weeks) were higher with vinorelbine compared with the control arm (30.6% versus 19.2%, p = 0.008). QoL data were collected in the trial but were limited due to poor patient compliance and use of a general rather than a specific prostate cancer QoL instrument; and showed no benefit with vinorelbine on either global QoL or functional subscales. More frequent severe neutropenia (26%), neutropenic infection (3%), anemia (6.5%) and constipation (3%) were observed with the addition of vinorelbine to hydrocortisone with or without aminoglutethimide.
Anthracenedione/anthracycline-based chemotherapy
Mitoxantrone
Mitoxantrone is an anthracenedione drug with mechanisms of activity similar to anthracyclines and a modest toxicity profile. Three RCTs compared mitoxantrone combined with low-dose corticosteroid to the same low-dose corticosteroid alone without placebo [69–71] (Table 4). In the largest trial, Kantoff et al [70] randomized 242 patients with metastatic HRPC who had undergone AAWD to either mitoxantrone plus hydrocortisone or hydrocortisone alone. The primary endpoint of the trial was overall survival. Tannock et al [71] compared mitoxantone plus prednisone to prednisone alone in 161 men with HRPC symptomatic with pain. Pain relief was the primary endpoint of that trial, defined by patient self-reported pain intensity and analgesic use as recorded in an analgesic diary. The analysis of overall survival was confounded by crossover to the mitoxantrone arm at the time of cancer progression in this trial. Berry et al [69] evaluated the same treatment regimens as Tannock et al in 120 men with asymptomatic HRPC using TTP as the primary endpoint.
All three RCTs reported overall survival results but none detected an improvement due to mitoxantrone (Table 5). Berry et al [69] and Kantoff et al [70] both reported longer median TTP with mitoxantrone versus control (8.1 versus 4.1 months [p = 0.018], and 3.7 versus 2.3 months [p = 0.02], respectively) (Table 5). Objective response rates were reported in two trials [69, 70] without differences observed (Table 6). All three trials reported PSA response rates, which were significantly higher with mitoxantrone in one trial [69] (Table 6).
In the only trial evaluating pain, Tannock et al [71] rigorously assessed palliative response through self-reported pain scores and analgesic consumption. In the mitoxantrone-prednisone treatment arm, 29% of patients had a two-point reduction in pain intensity (or complete elimination of pain) on the six-point McGill-Melzack Pain Questionnaire, maintained for three weeks apart without an increase in analgesic use, compared with 12% of patients treated with prednisone alone (p = 0.01). The median duration of pain response was 43 versus 18 weeks favoring mitoxantrone (p < 0.0001). An additional seven patients in each arm had a decrease of ≥ 50% in analgesic score without an increase in pain; thus, 38% of patients treated with mitoxantrone-prednisone had palliative benefit compared with 21% with prednisone alone (p = 0.025).
Two trials reported QoL data [70, 71]. Tannock et al [71] reported improved QoL with mitoxantrone-prednisone over prednisone alone in domains related to pain, physical activity or function, constipation, and mood with the Prostate Cancer Specific QoL Instrument and the European Organization for Research and Treatment of Cancer Quality of Life Core Questionnaire (EORTC QLQ-C30). Patients meeting the criteria for palliative response had improvements in most QoL domains, including overall well-being. Kantoff et al [70] also reported improved QoL favoring mitoxantrone in the Functional Living Index: Cancer (FLIC) emotional state and family disruption subscales.
All three trials reported toxicity. Grade 3–4 neutropenia occurred in 45% of cycles, and 63% and 48% of patients, respectively [69–71]. Neutropenic sepsis occurred in 6.9% and 2% of patients [69, 71]. Severe symptomatic non-hematological toxicities were rare; for example, severe nausea and vomiting occurred in 0.5% of cycles [71]. Cardiac dysfunction, either symptomatic or detected by reduced LVEF, was observed in 3.8% and 5% of patients [69, 71]. No toxic deaths were observed with mitoxantrone in any of those trials.
Doxorubicin and epirubicin
Anthracyclines are believed to exert their cytotoxic effects primarily through the inhibition of topoisomerase-II activity. Six RCTs examined anthracycline combinations [72–76, 88] (Table 4). Four performed in the pre-PSA era compared doxorubicin-based cytotoxic chemotherapy regimens to non-doxorubicin-based regimens or single agents [74–76] or compared combined versus sequential 5-flourouracil-doxorubicin-mitomycin-C (FAM) [73]. Two trials compared anthracyclines to EMP [72, 88]. The primary endpoints of those trials were tumor response and survival [73–76] and TTP [72].
Only one of five RCTs reporting overall survival data reported improved survival with chemotherapy [72–76, 88] (Table 5). Laurie et al [73] randomized 142 patients to either combination chemotherapy with FAM or sequential chemotherapy with the same drugs (mitomycin C followed by doxorubicin followed by 5-fluorouracil). Although response rates were similar between the two arms and hematological toxicity was greater with FAM, overall survival favored the combined FAM regimen (median survival, 8.7 versus 7.1 months, p = 0.025). Three trials provided comparative data on disease progression [72, 74, 75]; two reported on TTP [72, 74] and one reported on PFS [75] (Table 5). Improved TTP was detected with epirubicin and MPA compared with EMP (median, 7.6 versus 4.3 months, p = 0.013) [72].
Five trials reported on objective tumor response [73–76, 88] (Tables 6). Only two of the five trials provided statistical comparisons of those data; one detected no difference and the other reported higher response rates with combined doxorubicin-cyclophosphamide chemotherapy (32%) versus hydroxyurea (4%) that was of borderline statistical significance (p = 0.05) [76]. Two of the trials assessed a pain or palliative endpoint [76, 88]. Stephens et al [76] reported that symptomatic response rate (a composite endpoint comprising of worsening symptoms and analgesic use) was higher with doxorubicin plus cyclophosphamide compared with hydroxyurea (26% versus 13%, p = 0.048) but the duration of that response was not significantly different between the two groups (p = 0.62). The second trial [88] reported comparable rates of pain relief (undefined) among patients treated with epirubicin (49%), mitomycin C (48%), and EMP (42%). None of the six trials reported QoL data.
Other cytotoxic agents
Four trials studied other chemotherapy agents [75, 77, 78, 88] (Table 7). The National Prostatic Cancer Project (NPCP) randomized 189 men with clinically progressing HRPC to either single-agent cisplatin, methotrexate, or EMP [78]. In a successor trial, 180 patients were randomized to either single-agent methotrexate, combination cyclophosphamide-5-fluorouracil-cisplatin, or cyclophosphamide-doxorubicin [75]. Objective response by NPCP criteria was the primary endpoint of both trials. Newling et al [77] compared mitomycin C with EMP in 171 randomized patients with TTP and overall survival as primary endpoints. All three trials were completed during the pre-PSA era. Weissbach et al [88] randomized 175 patients to mitomycin C, epirubicin, or EMP.
All four trials reported data on overall survival [75, 77, 78, 88], but none reported differences between trial arms. Two trials reported on disease progression [75, 77] and no differences were detected. Improved time-to-treatment failure was reported with mitomycin C compared with epirubicin (p = 0.039) and EMP (p = 0.037) [88]. Trials reporting symptomatic or pain response identified no differences [78]. Three trials [75, 78, 88] reported tumor response data and no differences were observed. One trial collected QoL data, but it was of limited value due to missing data [77].
Non-cytotoxic agents
Five non-cytotoxic agents, including liarozole, suramin, atrasentan, prinomastat, and APC8015 have been investigated in RCTs in HRPC (Table 8) [79–83, 85–87]. Liarozole is thought able to promote the differentiation of malignant cells by increasing intracellular levels of retinoic acid. Debruyne et al [83] randomized 321 patients to either liarozole or cyproterone acetate. Suramin is a highly charged polysulfonated napthylurea with antineoplastic activity of uncertain mechanisms and adrenolytic effects. Two recent, large RCTs have studied suramin in men with HRPC. In a placebo-controlled trial, Small et al [82] studied the effects of suramin plus hydrocortisone to hydrocortisone alone in men with HRPC and pain requiring opioid analgesics. The primary endpoint was pain response. A subsequent RCT compared three different doses of suramin and evaluated PSA response as the primary endpoint [81]. Atrasentan is an orally bioavailable endothelin A antagonist. Two large RCTs have studied atrasentan in comparison to placebo in men with HRPC [79, 85]. The matrix metalloprotease inhibitor prinomastat has been combined with mitoxantrone-prednisone and compared with placebo [87]. APC8015 is a cellular therapy consisting of autologous peripheral blood mononuclear cells enriched for dendritic cells and pulsed with a prostatic acid phosphatase-GM-CSF construct. APC8015 has also been compared with placebo in men with HRPC [86].
Four of the seven trials of non-cytotoxic agents reported overall survival results, and none reported differences in overall survival between treatment arms (Table 9) [80–83, 87]. Reduced mortality was reported when liarozole was compared with cyproterone acetate after an adjustment for prognostic factors by Cox multivariate regression analysis (HR = 0.74 [95% CI, 0.56–0.99], p = 0.039) [83]. All eight trials reported on a disease-progression outcome; four trials reported TTP [79, 82, 85, 86], three reported on PFS [81, 83, 87] and one reported failure-free survival data [80] (Table 9). Of those trials, two detected statistically significant differences favoring the experimental treatment [80, 82]. TTP was improved with suramin-hydrocortisone compared with placebo-hydrocortisone (relative risk = 1.51 [95% CI, 1.22–1.85], two-sided p = 0.0003) [82], but was not affected by suramin schedule in another trial [81]. Tumor response data were reported in three trials [80–82] (Table 10), of which only one detected significant differences between trial arms [80]. PSA response rates were reported in seven trials [79, 81–83, 85–87]; four of those detected statistically significantly higher response rates with the experimental therapy [79, 82, 83, 85] (Table 10).
Three trials reported pain or symptomatic response data [80, 82, 83, 86]. The mean best change in pain and analgesic use score compared with baseline was improved with liarozole compared with cyproterone acetate (mean reduction 0.4 versus 0.2, p = 0.026) [83]. Small et al [82] assessed palliative response to suramin with self-reported pain scores and opioid analgesic use using two methods. For the first method, average worst pain scores (during the previous 24 hours) measured with the Brief Pain Inventory (BPI) and opioid analgesic consumption were assessed in each treatment group and compared with baseline at six weeks and at the end of treatment. Suramin was superior to placebo for pain reduction at both six weeks and the end of study; no statistically significant differences in narcotic analgesic consumption were observed. For the second method, pain response was measured and defined either by a three-point reduction (or complete elimination) of worst pain on the BPI (maintained for at least three weeks) with a <16% increase in opioid analgesic use or by a ≥ 33% (minimum 5 mg) reduction in opioid analgesic use with a two-point or less increase in pain. More patients in the suramin group had pain response compared with placebo (43% versus 28%, p = 0.001), and the duration of pain response was significantly higher (median 240 versus 69 days, two-sided p = 0.0027). Performance status measured by the Revised Rand Functional Limitations Scale was not improved with suramin compared with placebo. Three trials assessed QoL outcomes [79, 82, 83] and none reported differences.
Liarozole was associated with increased rates of skin toxicity, nausea and vomiting, and fatigue [83]. In comparison to placebo, suramin was associated with more frequent mild to moderate rash (57% versus 13%) and severe edema and anemia (both <5%) [82]. Higher rates of severe toxicities were seen with high-dose suramin, including neutropenia, anorexia, cardiac dysrhythmias, and neuromotor toxicity [81]. Prinomastat was associated with increased rates of mild to moderate musculoskeletal effects including arthralgia, joint stiffness and swelling, and, rarely, contracture compared with placebo [87]. Atrasentan was associated with increased rates of mild to moderate peripheral edema, rhinitis, headache, hypotension, anemia, and weight gain compared with placebo [79].
Discussion
The diagnosis and clinical management of HRPC has undergone radical changes over the past decade, along with the design and methodology of clinical trials. A "stage migration" has occurred due to the ability of PSA testing to detect biochemical evidence of androgen independence before other clinical symptoms or signs become apparent. In the pre-PSA era, men with HRPC enrolled in RCTs were often symptomatic and extensively pre-treated with palliative radiotherapy. Androgen levels influence tumor growth in HRPC, and differences in or lack of control of ADT used in trial subjects might affect outcomes. For example, luteinizing hormone-releasing hormone (LHRH)-agonist use has become much more prevalent over the past decade, replacing estrogens and reducing the use of bilateral orchiectomy. As well, the AAWD syndrome has been identified as a potential confounder of clinical and biochemical response in HRPC [89]. Withdrawal of oral antiandrogens and maintenance of ADT were required for entry onto the largest clinical trials [63, 64, 70, 71] discussed in this review. Generalizability, changes in use of androgen deprivation therapy, and the validity of trial endpoints need to be considered in the interpretation and weighting of the evidence provided by clinical trials in HRPC.
Typically, men with HRPC have skeletal metastases that cannot be conventionally assessed for objective response to anticancer therapy. As a result, drug trials have used a number of different primary endpoints. The identification of benefits from non-hormonal drug therapy only became clear over the past 15 years with the emergence first of validated symptom and quality of life instruments, then the availability of PSA as a tumor marker, and finally the ability to conduct randomized trials large enough to adequately assess survival benefits. As the purpose of this review was to inform clinical practice, endpoints unequivocally associated with patient benefit or harm were emphasized, as were RCTs of sufficient power to assess these.
Early RCTs studying several single-agent and combination chemotherapy regimens compared with other single-agent chemotherapy controls showed evidence of modest anti-tumor activity generally accompanied by increased toxicity. Interpretation of the results of these trials was limited by their sample sizes and lack of validated psychometric tools to ascertain palliative treatment benefits. Tannock et al [71] established mitoxantrone-prednisone as a standard palliative therapy for men with HRPC symptomatic with pain. Two trials [69, 70] subsequently confirmed that mitoxantrone also improved TTP compared to initial corticosteroid therapy alone. The lack of toxicity in those trials was notable, with no toxic deaths and few serious hematological and non-hematological side effects. Cardiomyopathy was observed in ≥ 5% of patients in those trials. Improved PFS was also associated with vinorelbine, along with a modest benefit in clinical benefit response [65]. A number of agents with novel mechanisms of anti-tumor activity have been studied in HRPC; and although activity and some benefits have been observed, data from RCTs has not yet established any of these agents as standard therapeutic options for HRPC. The strategy of adding EMP to chemotherapy has also been explored, and PSA response and disease control appear modestly improved [64, 68, 87]. However, overall survival is not clearly increased, and EMP is associated with adverse effects that include thrombosis and cardiovascular toxicity [61, 63, 65, 84].
Improvement in overall survival has been reported with docetaxel given every three weeks in comparison with mitoxantrone-prednisone in two large, well-conducted RCTs [63, 64]. Docetaxel-prednisone given on a weekly schedule was not clearly associated with improved overall survival [64]. These trials also reported evidence of improved PFS, palliative response, and/or objective response with all the docetaxel regimens studied. Docetaxel-prednisone (without estramustine) was associated with more frequent mild toxicities, similar rates of serious toxicities, and better QoL than mitoxantrone-prednisone. Based on this evidence, docetaxel given every third week with either daily prednisone or EMP appears to be the most effective drug treatment tested in RCTs for men with HRPC and metastases; however, indirect comparison suggests the latter regimen may be associated with more frequent and severe toxicities than the former.
Conclusion
Docetaxel-based chemotherapy given every three weeks was the only treatment that demonstrated an overall survival benefit in men with HRPC. Most men receiving docetaxel had metastases, so the timing of docetaxel therapy in men without metastases should not only be carefully considered but also studied further in clinical trials. Expectant management, trials of secondary hormonal manipulations, and/or participation in clinical trials of investigational agents before chemotherapy are also reasonable alternatives for many men with HRPC on an individualized basis. In the largest randomized trials, men continued on gonadal androgen suppression and discontinued the use of oral antiandrogens, and these maneuvers are recommended for men planned to receive chemotherapy. Use of EMP in combination with other cytotoxic agents is probably not worthwhile due to the increased risk of clinically important toxicities without clear evidence of improved survival or palliation. Mitoxantrone-prednisone and weekly docetaxel-prednisone are associated with symptom palliation and improved disease control without improved overall survival, and can be considered clinical alternatives to docetaxel given every three weeks.
References
Jemal A, Clegg LX, Ward E, Ries LAG, Wu X, Jamison PM: Annual report to the Nation on the status of cancer, 1975-2001, with a special feature regarding survival. Cancer. 2004, 101: 3-27. 10.1002/cncr.20288.
Sharifi N, Dahut WL, Steinberg SM, Figg WD, Tarassoff C, Arlen P, Gulley JL: A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU Int. 2005, 96: 985-989. 10.1111/j.1464-410X.2005.05798.x.
Newling DW, Dennis L, Vermylen K: Orchiectomy versus goserelin and flutamide in the treatment of newly diagnosed metastatic prostate cancer. Cancer. 1993, 72: 3793-3798. 10.1002/1097-0142(19931215)72:12+<3793::AID-CNCR2820721706>3.0.CO;2-U.
Coleman RE: Skeletal complications of malignancy. Cancer. 1997, 80: 1588-1594. 10.1002/(SICI)1097-0142(19971015)80:8+<1588::AID-CNCR9>3.0.CO;2-G.
Oh WK, Kantoff PW: Management of hormone refractory prostate cancer: current standards and future prospects. J Urol. 1998, 160: 1220-1229. 10.1097/00005392-199810000-00003.
Browman GP, Levine MN, Mohide EA, Hayward RSA, Pritchard KI, Gafni A, Laupacis A: The practice guidelines development cycle: a conceptual tool for practice guidelines development and implementation. J Clin Oncol. 1995, 13: 502-512.
Sterne JAC, Egger M, Smith GD: Investigating and dealing with publication and other biases. Systematic Reviews in Health Care: Meta-analysis in Context. Edited by: Egger M, Smith GD, Altman D. 2001, London: BMJ Publishing Group, 189-208.
Scott WW, Gibbons RP, Johnson DE, Prout GR, Schmidt JD, Saroff J, Murphy GP: The continued evaluation of the effects of chemotherapy in patients with advanced carcinoma of the prostate. Trans Am Assoc Genitourin Surg. 1977, 68: 24-28.
Eagan RT, Hahn RG, Myers RP: Adriamycin (NSC-123127) versus 5-fluorouracil (NSC-19893) and cyclophosphamide (NSC-26271) in the treatment of metastatic prostate cancer. Cancer Treatment Reports. 1976, 60: 115-117.
Kvols LK, Eagan RT, Myers RP: Evaluation of melphalan, ICRF-159, and hydroxyurea in metastatic prostate cancer: a preliminary report. Cancer Treatment Reports. 1977, 61: 311-312.
Murphy GP, Gibbons RP, Johnson DE, Loening SA, Prout GR, Schmidt JD, Bross DS, Chu TM, Gaeta JF, Saroff J, Scott WW: A comparison of estramustine phosphate and streptozotocin in patients with advanced prostatic carcinoma who have had extensive irradiation. J Urol. 1977, 118: 288-291.
Tejada F, Eisenberger MA, Broder LA, Cohen MH, Simon R: 5-fluorouracil versus CCNU in the treatment of metastatic prostatic cancer. Cancer Treatment Reports. 1977, 61: 1589-1590.
Chlebowski RT, Hestorff R, Sardoff L, Weiner J, Bateman JR: Cyclophosphamide (NSC 26271) versus the combination of adriamycin (NSC 123127), 5-fluorouracil (NSC 19893), and cyclophosphamide in the treatment of metastatic prostatic cancer: a randomized trial. Cancer. 1978, 42: 2546-2552. 10.1002/1097-0142(197812)42:6<2546::AID-CNCR2820420607>3.0.CO;2-C.
Schmidt JD, Scott WW, Gibbons RP, Johnson DE, Prout GR, Loening SA, Soloway MS, Chu TM, Gaeta JF, Slack NH, Saroff J, Murphy GP: Comparison of procarbazine, imidazole-carboxamide and cyclophosphamide in relapsing patients with advanced carcinoma of the prostate. J Urol. 1979, 121: 185-189.
Loening SA, Scott WW, deKernion J, Gibbons RP, Johnson DE, Pontes JE, Prout GR, Schmidt JD, Soloway MS, Chu TM, Gaeta JF, Slack NH, Murphy GP: A comparison of hydroxyurea, methyl-chloroethyl-cyclohexy-nitrosourea and cyclophosphamide in patients with advanced carcinoma of the prostate. J Urol. 1981, 125: 812-816.
Muss HB, Howard V, Richards F, White DR, Jackson DV, Cooper MR, Stuart JJ, Resnick MI, Brodkin R, Spurr CL: Cyclophosphamide versus cyclophosphamide, methotrexate, and 5-fluorouracil in advanced prostatic cancer: a randomized trial. Cancer. 1981, 47: 1949-1953. 10.1002/1097-0142(19810415)47:8<1949::AID-CNCR2820470806>3.0.CO;2-3.
Smalley RV, Bartolucci AA, Hemstreet G, Hester M: A phase II evaluation of a 3-drug combination of cyclophosphamide, doxorubicin and 5-fluorouracil and of 5-fluorouracil in patients with advanced bladder carcinoma or stage D prostatic carcinoma. J Urol. 1981, 125: 191-195.
Soloway MS, Dekernion JB, Gibbons RP, Johnson DE, Loening SA, Pontes JE, Prout GR, Schmidt JD, Scott WW, Chu TM, Gaeta JF, Slack NH, Murphy GP: Comparison of estramustine phosphate and vincristine alone or in combination for patients with advanced, hormone refractory, previously irradiated carcinoma of the prostate. J Urol. 1981, 125: 664-667.
Herr HW: Cyclophosphamide, methotrexate and 5-fluorouracil combination chemotherapy versus chloroethyl-cyclohexy-nitrosourea in the treatment of metastatic prostatic cancer. J Urol. 1982, 127: 462-465.
DeWys WD, Begg CB, Brodovsky H, Creech R, Khandekar J: A comparative clinical trial of adriamycin and 5-fluorouracil in advanced prostatic cancer: prognostic factors and response. Prostate. 1983, 4: 1-11.
Soloway MS, Beckley S, Brady MF, Chu TM, Dekernion JB, Dhabuwala C, Gaeta JF, Gibbons RP, Loening SA, McKiel CF, McLeod DG, Pontes JE, Prout GR, Scardino PT, Schlegel JU, Schmidt JD, Scott WW, Slack NH, Murphy GP: A comparison of estramustine phosphate versus cis-platinum alone versus estramustine phosphate plus cis-platinum in patients with advanced hormone refractory prostate cancer who have had extensive irradiation to the pelvis or lumbosacral area. J Urol. 1983, 129: 56-61.
Kasimis BS, Miller JB, Kaneshiro CA, Forbes KA, Moran EM, Metter GE: Cyclophosphamide versus 5-fluorouracil, doxorubicin, and mitomycin C (FAM') in the treatment of hormone-resistant metastatic carcinoma of the prostate: a preliminary report of a randomized trial. J Clin Oncol. 1985, 3: 385-392.
Page JP, Levi JA, Woods RL, Tattersall MN, Fox RM, Coates AS: Randomized trial of combination chemotherapy in hormone-resistant metastatic prostate carcinoma. Cancer Treatment Reports. 1985, 69: 105-107.
Torti FM, Shortliffe LD, Carter SK, Hannigan JF, Aston D, Lum BL, Williams RD, Spaulding JT, Freiha FS: A randomized study of doxorubicin versus doxorubicin plus cisplatin in endocrine-unresponsive metastatic prostatic carcinoma. Cancer. 1985, 56: 2580-2586. 10.1002/1097-0142(19851201)56:11<2580::AID-CNCR2820561108>3.0.CO;2-W.
Benson RC, Cummings K: Estramustine phosphate vs. diethylstilbestrol in the treatment of stage D prostate cancer. Prog Clin Biol Res. 1989, 303: 177-186.
Graham SD, Walker A, Cox EB, Laszlo J, Berry WR, Paulson DF: Value of cyclophosphamide or melphalan as combined chemotherapy in hormonally unresponsive prostatic carcinoma. Urology. 1986, 28: 404-408. 10.1016/0090-4295(86)90074-9.
Akaza H, Isurugi K, Oishi Y, Kitajima K, Sawamura Y, Baba S, Yoshida K, Otani M, Harada M, Gunji A: A prospective, randomized controlled study on the treatment of stage C and stage D prostatic cancer with estracyt in combination with other chemotherapeutic agents. Jpn J Clin Oncol. 1988, 18: 343-355.
Kitahara S, Fukui I, Higashi Y, Kihara K, Takeuchi S, Oshima H, Negishi T, Hosoda K, Kawai T, Ikegami S: [A randomized trial of chemotherapy of advanced prostatic cancer with ifosfamide alone versus ifosfamide, 5-fluorouracil and cisplatin (preliminary report)] [Japanese]. Nippon Gan Chiryo Gakkai Shi. 1988, 23: 2507-2513.
Manni A, Bartholomew M, Caplan R, Boucher A, Santen R, Lipton A, Harvey H, Simmonds M, White-Hershey D, Gordon R: Androgen priming and chemotherapy in advanced prostate cancer: evaluation of determinants of clinical outcome. J Clin Oncol. 1988, 6: 1456-1466.
McLeod DG, Murphy GP, Priore R: Comparison of megace, stilphostrol, megace plus DES, or streptozotocin in metastatic prostatic cancer in patients with hormonal failure and prior radiotherapy. Urology. 1988, 32: 431-436. 10.1016/0090-4295(88)90419-0.
Papadopoulos I, Wand H: [Reducing the side effects of aggressive chemotherapy (cisplatin and epirubicin) with xenogenic peptides (factor AF2) in patients with hormone refractory metastatic prostate cancer. A prospective, randomized study] [German]. Onkologie. 1989, 12 (Suppl 3): 26-31.
Ruff P, Derman DP, Weaving A, Bezwoda WR: Sequential hormonal therapy and sequential hormonal and chemotherapy for advanced prostatic cancer. Oncology. 1989, 46: 288-292.
Shafik A: Anal submucosal injection: a new route for drug administration in pelvic malignancies. V. Advanced prostatic cancer: results of methotrexate treatment using the anal route – preliminary study. Eur Urol. 1990, 18: 132-136.
Tveter KJ, Hagen S, Holme I, Klepp O, Kloster SE, Muri O, Poulsen C, Sander S, Sivertsen S, Urnes T: A randomized study on hormone-resistant prostatic cancer: estramustine phosphate versus low dose epirubicin with or without medroxyprogesterone acetate. A Norwegian multicenter study. Scand J Urol Nephrol. 1990, 24: 243-247.
Elomaa I, Kellokumpu-Lehtinen P, Rannikko S, Alfthan O: Hormone-resistant metastatic prostate cancer. Comparisons between estramustine phosphate and low-dose epirubicin treatments. Eur Urol. 1991, 19: 12-15.
Rangel C, Matzkin H, Soloway MS: Experience with weekly doxorubicin (adriamycin) in hormone-refractory stage D2 prostate cancer. Urology. 1992, 39: 577-582. 10.1016/0090-4295(92)90023-P.
Francini G, Petrioli R, Manganelli A, Cintorino M, Marsili S, Aquino A, Mondillo S: Weekly chemotherapy in advanced prostatic cancer. Br J Cancer. 1993, 67: 1430-1436.
Daliani DD, Eisenberg PD, Weems J, Lord R, Fueger R, Logothetis CJ: The results of a phase II randomized trial comparing 5-fluorouracil and 5-fluorouracil plus alpha-interferon: observations on the design of clinical trials for androgen-independent prostate cancer. J Urol. 1995, 153: 1587-1591. 10.1097/00005392-199505000-00064.
Breul J, Jakse G, Forster G, Lampel A, Rohani A, Hartung R: 5-fluorouracil versus folinic acid and 5-fluorouracil in advanced, hormone-resistant prostate cancer: a prospective randomized pilot trial. Eur Urol. 1997, 32: 280-283.
Brune D: Phase III trial of pirarubicin versus palliative treatment in hormone-refractory prostatic cancer [abstract]. Proc Am Soc Clin Oncol. 1998, 17: 316a-
Van Andel G, Kurth KH, Rietbroek RL, Van de Velde-Muusers JA: Quality of life assessment in patients with hormone-resistant prostate cancer treated with epirubicin or with epirubicin plus medroxy progesterone acetate – Is it feasible?. Eur Urol. 2000, 38: 259-264. 10.1159/000020291.
Dahut W, Arlen P, Gulley J, Parker C, Fedenko K, Noone M, Hamilton M: A randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer [abstract]. Proc Am Soc Clin Oncol. 2002, 21: 183a-
Figg WD, Dahut W, Duray P, Hamilton M, Tompkins A, Steinberg SM, Jones E, Premkumar A, Linehan WM, Floeter MK, Chen CC, Dixon S, Kohler DR, Kruger EA, Gubish E, Pluda JM, Reed E: A randomized phase II trial of thalidomide, an angiogenesis inhibitor, in patients with androgen-independent prostate cancer. Clin Cancer Res. 2001, 7: 1888-1893.
Millikan R, Baez L, Banerjee T, Wade J, Edwards K, Winn R, Smith TL, Logothetis C: Randomized phase 2 trial of ketoconazole and ketoconazole/doxorubicin in androgen independent prostate cancer. Urol Oncol. 2001, 6: 111-115. 10.1016/S1078-1439(00)00123-X.
Hervonen P, Lehtinen T, Tammela TL, Kellokumpu-Lehtinen P: A randomised dose-finding phase II study on ifosfamide in metastatic hormone-refractory prostate cancer (HRPC). J Exp Clin Cancer Res. 2002, 21: 177-180.
Tombal B, Tourani J-M, Fizazi K, Culine S, Lotz J-P, Ould Kaci M, Coppola F: Randomized phase II trial of irofulven (IROF) with or without prednisone in hormone-refractory prostate cancer (HRPC) patients (pts) [abstract]. Proc Am Soc Clin Oncol. 2003, 22: 407-
Tolcher AW, Reyno L, Venner PM, Ernst SD, Moore M, Geary RS, Chi K, Hall S, Walsh W, Dorr A, Eisenhauer E: A randomized phase II and pharmacokinetic study of the antisense oligonucleotides ISIS 3521 and ISIS 5132 in patients with hormone-refractory prostate cancer. Clin Cancer Res. 2002, 8: 2530-2535.
Oudard S, Beuzeboc P, Dourthe LM, Voog E, Hardy-Bessard AC, Coscas I, Chauvet B: Preliminary results of a phase II randomized trial of docetaxel (D), estramustine (E), and prednisone (P) – two schedules – versus mitoxantrone (M) and prednisone in patients (pts) with hormone refractory prostate cancer (HRPC) [abstract]. Proc Am Soc Clin Oncol. 2002, 21: 177a-
Droz JP, Muracciole X, Mottet N, Ould KM, Vannetzel JM, Albin N, Culine S, Rodier JM, Misset JL, Mackenzie S, Cvitkovic E, Benoit G: Phase II study of oxaliplatin versus oxaliplatin combined with infusional 5-fluorouracil in hormone refractory metastatic prostate cancer patients. Ann Oncol. 2003, 14: 1291-1298. 10.1093/annonc/mdg342.
Millikan R, Thall PF, Lee SJ, Jones D, Cannon MW, Kuebler JP, Wade J, Logothetis CJ: Randomized, multicenter, phase II trial of two multicomponent regimens in androgen-independent prostate cancer. J Clin Oncol. 2003, 21: 878-883. 10.1200/JCO.2003.04.057.
Salimichokami M: Combining angiogenesis inhibitors with cytotoxic chemotherapy enhances PSA response in hormone refractory prostate cancer (HRPC), a randomized study of weekly docetaxel alone or in combination with thalidomide [abstract]. Proc Am Soc Clin Oncol. 2003, 22: 429-
Sternberg CN, Hetherington JW, Paluchowska B, Slee PH, Collette L, Debois M, Zurlo A: Randomized phase III trial of a new oral platinum, satraplatin (JM-216) plus prednisone or prednisone alone in patients with hormone refractory prostate cancer [abstract]. Proc Am Soc Clin Oncol. 2003, 22: 395-
Van Andel G, Fernandez de Moral P, Caris CT, Carpentier P, Wils J, de Bruin MJ, Witjes JA, Debruyne FM, Witjes WP: A randomized study comparing epirubicin in a 4-weekly versus a weekly intravenous regimen in patients with metastatic, hormone resistant, prostatic carcinoma: effects on health related quality of life. World J Urol. 2003, 21: 177-182. 10.1007/s00345-003-0342-3.
Albrecht W, Van Poppel H, Horenblas S, Mickisch G, Horwich A, Serretta V, Casetta G, Marechal JM, Jones WG, Kalman S, Sylvester R: Randomized phase II trial assessing estramustine and vinblastine combination chemotherapy vs estramustine alone in patients with progressive hormone-escaped metastatic prostate cancer. Br J Cancer. 2004, 90: 100-105. 10.1038/sj.bjc.6601468.
Dimopoulos MA, Kiamouris C, Gika D, Deliveliotis C, Giannopoulos A, Zervas A, Alamanis C, Constantinidis C, Koutsilieris M: Combination of LHRH analog with somatostatin analog and dexamethasone versus chemotherapy in hormone-refractory prostate cancer: a randomized phase II study. Urology. 2004, 63: 120-125. 10.1016/j.urology.2003.08.041.
Birch R, Kalman L, Holt L, Graham B, Wheeler B, Schwartzberg L: Randomized phase IIb trial comparing two schedules of docetaxel (D) plus estramustine (E) for metastatic hormone refractory prostate cancer (HRPC) [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 411-
Eymard J-C, Joly F, Priou F, Zannetti A, Ravaud A, Kerbrat P, Mousseau M: Phase II randomized trial of docetaxel plus estramustine (DE) versus docetaxel (D) in patients (pts) with hormone-refractory prostate cancer (HRPC): a final report [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 406-
Kelly WK, Galsky MD, Small EJ, Oh W, Chen I, Smith D, Martone L: Multi-institutional trial of the epothiline B analogue BMS-247550 with or without estramustine phosphate (EMP) in patients with progressive castrate-metastatic prostate cancer (PCMPC): updated results [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 383-
Lara PN, Longmate J, Stadler W, Quinn DI, Twardowski P, Martel CI, Vogelzang NJ: Angiogenesis inhibition in metastatic hormone refractory prostate cancer (HRPC): a randomized trial of two doses of the matrix metalloproteinase inhibitor (MMPI) BMS-275291 [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 417-
Millikan R, Thall P, Pagliaro L, Williams D, Brown M, Logothetis C: Randomized, adaptive, phase II selection trial of four chemotherapy regimens in androgen independent prostate cancer (AIPC) [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 395-
Hudes G, Einhorn L, Ross E, Balsham A, Loehrer P, Ramsey H, Sprandio J, Entmacher M, Dugan W, Ansari R, Monaco F, Hanna M, Roth B: Vinblastine versus vinblastine plus oral estramustine phosphate for patients with hormone-refractory prostate cancer: A Hoosier Oncology Group and Fox Chase Network phase III trial. J Clin Oncol. 1999, 17: 3160-3166.
Abratt RP, Brune D, Dimopoulos MA, Kliment J, Breza J, Selvaggi FP, Beuzeboc P: Randomised phase III study of intravenous vinorelbine plus hormone therapy versus hormone therapy alone in hormone-refractory prostate cancer. Ann Oncol. 2004, 15: 1613-1621. 10.1093/annonc/mdh429.
Petrylak DP, Tangen CM, Maha PH, Hussain HA, Lara PN, Jones JA, Taplin ME: Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004, 351: 1513-1520. 10.1056/NEJMoa041318.
Tannock IF, De Wit R, Berry WR, Horti J, Pluzanska A, Chi K, Oudard S: Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004, 351: 1502-1512. 10.1056/NEJMoa040720.
Iversen P, Rasmussen F, Asmussen C, Christensen IJ, Eickhoff J, Klarskov P, Larsen E, Mogensen P, Mommsen S, Rosenkilde P: Estramustine phosphate versus placebo as second line treatment after orchiectomy in patients with metastatic prostate cancer: DAPROCA study 9002. Danish Prostatic Cancer Group. J Urol. 1997, 157: 929-934. 10.1016/S0022-5347(01)65086-9.
Johansson JE, Andersson SO, Holmberg L: High-dose medroxyprogesterone acetate versus estramustine in therapy-resistant prostatic cancer: a randomised study. Br J Urol. 1991, 68: 67-73.
de Kernion JN, Murphy GP, Priore R: Comparison of flutamide and Emcyt in hormone-refractory metastatic prostatic cancer. Urology. 1988, 31: 312-317. 10.1016/0090-4295(88)90088-X.
Murphy GP, Gibbons RP, Johnson DE, Prout GR, Schmidt JD, Soloway MS, Loening SA, Chu TM, Gaeta JF, Saroff J, Wajsman Z, Slack N, Scott WW: The use of estramustine and prednimustine versus prednimustine alone in advanced metastatic prostatic cancer patients who have received prior irradiation. Trans Am Assoc Genitourin Surg. 1979, 70: 69-71.
Berry W, Dakhil S, Modiano M, Gregurich M, Asmar L: Phase III study of mitoxantrone plus low dose prednisone versus low dose prednisone alone in patients with asymptomatic hormone refractory prostate cancer. J Urol. 2002, 168: 2439-2443. 10.1016/S0022-5347(05)64163-8.
Kantoff PW, Halabi S, Conaway M, Picus J, Kirshner J, Hars V, Trump D, Winer EP, Vogelzang NJ: Hydrocortisone with or without mitoxantrone in men with hormone-refractory prostate cancer: results of the Cancer and Leukemia Group B 9182 study. J Clin Oncol. 1999, 17: 2506-2513.
Tannock IF, Osoba D, Stockler MR, Ernst DS, Neville AJ, Moore MJ, Armitage GR, Wilson JJ, Venner PM, Coppin CM, Murphy KC: Chemotherapy with mitoxantrone plus prednisone or prednisone alone for symptomatic hormone-resistant prostate cancer: a Canadian randomized trial with palliative end points. J Clin Oncol. 1996, 14: 1756-1764.
Anderström C, Eddeland A, Folmerz P, Hansson R, Milles S, Zachrisson B: Epirubicin and medroxyprogesterone acetate versus estramustine phosphate in hormone-resistant prostatic cancer: a prospective randomized study. Eur Urol. 1995, 27: 301-305.
Laurie JA, Hahn RG, Therneau TM, Patel SR, Mailliard JA, Windschitl HE, Twito DI, Morton RF, Krook JE: Chemotherapy for hormonally refractory advanced prostate carcinoma. A comparison of combined versus sequential treatment with mitomycin C, doxorubicin, and 5-fluorouracil. Cancer. 1992, 69: 1440-1444. 10.1002/1097-0142(19920315)69:6<1440::AID-CNCR2820690622>3.0.CO;2-9.
Saxman S, Ansari R, Drasga R, Miller M, Wheeler B, McClean J, Einhorn L: Phase III trial of cyclophosphamide versus cyclophosphamide, doxorubicin, and methotrexate in hormone-refractory prostatic cancer. A Hoosier Oncology Group Study. Cancer. 1992, 70: 2488-2492. 10.1002/1097-0142(19921115)70:10<2488::AID-CNCR2820701016>3.0.CO;2-9.
Murphy GP, Priore RL, Scardino PT: Hormone-refractory metastatic prostatic cancer treated with methotrexate, cyclophosphamide plus adriamycin, cis-platinum plus 5-fluorouracil plus cyclophosphamide. National Prostatic Cancer Project randomized trial. Urology. 1988, 32: 33-40. 10.1016/0090-4295(88)90450-5.
Stephens RL, Vaughn C, Lane M, Costanzi J, O'Bryan R, Balcerzak SP, Levin H, Frank J, Coltman CA: Adriamycin and cyclophosphamide versus hydroxyurea in advanced prostatic cancer. A randomized Southwest Oncology Group study. Cancer. 1984, 53: 406-410. 10.1002/1097-0142(19840201)53:3<406::AID-CNCR2820530307>3.0.CO;2-3.
Newling DW, Fossa SD, Tunn UW, Kurth KH, de Pauw M, Sylvester R: Mitomycin C versus estramustine in the treatment of hormone resistant metastatic prostate cancer: the final analysis of the European Organization for Research and Treatment of Cancer, Genitourinary Group prospective randomized phase III study (30865). J Urol. 1993, 150: 1840-1844.
Loening SA, Beckley S, Brady MF, Chu TM, Dekernion JB, Dhabuwala C, Gaeta JF, Gibbons RP, McKiel CF, McLeod DG, Pontes JE, Prout GR, Scardino PT, Schlegel JU, Schmidt JD, Scott WW, Slack NH, Soloway MS, Murphy GP: Comparison of estramustine phosphate, methotrexate and cis-platinum in patients with advanced, hormone refractory prostate cancer. J Urol. 1983, 129: 1001-1006.
Carducci MA, Padley RJ, Breul J, Vogelzang NJ, Zonnenberg BA, Daliani DD, Schulman CC, Nabulsi AA, Humerickhouse RA, Weinberg MA, Schmitt JL, Nelson JB: Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: a randomized, phase II, placebo-controlled trial. J Clin Oncol. 2003, 21: 679-689. 10.1200/JCO.2003.04.176.
Leaf AN, Propert K, Corcoran C, Catalano PJ, Trump DL, Harris JE, Davis TE: Phase III study of combined chemohormonal therapy in metastatic prostate cancer (ECOG 3882): an Eastern Cooperative Oncology Group study. Med Oncol. 2003, 20: 137-146. 10.1385/MO:20:2:137.
Small EJ, Halabi S, Ratain MJ, Rosner G, Stadler W, Palchak D, Marshall E, Rago R, Hars V, Wilding G, Petrylak D, Vogelzang NJ: Randomized study of three different doses of suramin administered with a fixed dosing schedule in patients with advanced prostate cancer: results of Intergroup Cancer and Leukemia Group B 9480. J Clin Oncol. 0159, 20: 3369-3375. 10.1200/JCO.2002.10.022.
Small EJ, Meyer M, Marshall ME, Reyno LM, Meyers FJ, Natale RB, Lenehan PF, Chen L, Slichenmyer WJ, Eisenberger M: Suramin therapy for patients with symptomatic hormone-refractory prostate cancer: results of a randomized phase III trial comparing suramin plus hydrocortisone to placebo plus hydrocortisone. J Clin Oncol. 2000, 18: 1440-1450.
Debruyne FJ, Murray R, Fradet Y, Johansson JE, Tyrrell C, Boccardo F, Denis L, Marberger JM, Brune D, Rassweiler J, Vangeneugden T, Bruynseels J, Janssens M, De Porre P: Liarozole – a novel treatment approach for advanced prostate cancer: results of a large randomized trial versus cyproterone acetate. Liarozole Study Group. Urology. 1998, 52: 72-81. 10.1016/S0090-4295(98)00129-0.
Berry W, Gregurich M, Dakhil S, Hathorn J, Asmar L: Phase II randomized trial of weekly paclitaxel (Taxol®) with or without estramustine phosphate in patients with symptomatic, hormone-refractory, metastatic carcinoma of the prostate (HRMCP) [abstract]. Proc Am Soc Clin Oncol. 2001, 20: 175a-
Carducci M, Nelson JB, Saad F, Schulman C, Dearnaley DP, Sleep DJ, Hulting SM: Effects of atrasentan on disease progression and biological markers in men with metastatic hormone-refractory prostate cancer: phase 3 study [abstract]. Proc Am Soc Clin Oncol. 2004, 23: 383-
Small E, Rini B, Higano CS, Redfern C, Neumunaitis J, Valone F, Kylstra J: A randomized, placebo-controlled phase III trial of APC8015 in patients with androgen-independent prostate cancer (AIPCa) [abstract]. Proc Am Soc Clin Oncol. 2003, 22: 382-
Ahmann FR, Saad F, Mercier R, Huddart RA, Roberts JT, Collier M, Bettencourt L: Interim results of a phase III study of the matrix metalloprotease inhibitor prinomastat in patients having metastatic hormone refractory prostate cancer (HRPC) [abstract]. Proc Am Soc Clin Oncol. 2001, 20: 174a-
Weissbach L, Vogler H, Hofmann E, Knorr B: Palliative monochemotherapie des hormonrefrakt symptomat PCA's – ergebnisse einer prospektiven, randomisierten multizenterstudie. Urologe A. 1998, 37: 5.1-
Scher HI, Kelly WK: Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol. 1993, 11: 1566-1572.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1471-2407/6/112/prepub
Acknowledgements
The authors wish to thank Roanne Segal and Christina Canil, as well as the other members of the CCOPEBC Genitourinary Cancer Disease Site Group, and oncology practitioners in Ontario who reviewed the draft practice guideline from which this review is derived. This project was supported by Cancer Care Ontario and the Ontario Ministry of Health and Long-Term Care.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
EW has received honoraria from Sanofi-Avenitis. TW has no competing interests. SB has served as a consultant and received honoraria and research funding from Aventis. DSE has served as a consultant and received honoraria from Novartis, and has received honoraria and research funding from Aventis. SH has no competing interests. HL has received honoraria from Aventis.
Authors' contributions
EW was the lead author responsible for the conception and design of the systematic review, screening of citations, data auditing, analysis and interpretation, and drafting and editing of the manuscript. TW conducted literature searches, data extraction, and drafted and edited the manuscript. SB, DSE, and SH also screened citations; and SB, DSE, SH and HL reviewed all drafts of the manuscript and made major contributions to the interpretation of the evidence and manuscript revisions. All authors read and approved the final manuscript.
Electronic supplementary material
12885_2005_464_MOESM1_ESM.doc
Additional file 1: Appendix of ineligible randomized trialsThe file is a word document and describes the randomized trials that were excluded from the systematic review because they did not meet the eligibility criteria. (DOC 208 KB)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Winquist, E., Waldron, T., Berry, S. et al. Non-hormonal systemic therapy in men with hormone-refractory prostate cancer and metastases: a systematic review from the Cancer Care Ontario Program in Evidence-based Care's Genitourinary Cancer Disease Site Group. BMC Cancer 6, 112 (2006). https://doi.org/10.1186/1471-2407-6-112
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1471-2407-6-112